Primární lymfedém
- Abnormal accumulation of protein-rich fluid in the interstitial space
- Rare genetic condition
- Variable in age at onset, expressivity, and penetrance [1]
Dle dědičnosti
Autosomal dominant
- Single copy of an abnormal gene is necessary for the appearance of the disease
- Can be inherited from either parent
- Can be the result of a new mutation
- Risk of passing the abnormal gene from affected parent to offspring
- 50 percent for each pregnancy
- Regardless of the sex of the resulting child [3]
Autosomal recessive
Hereditary Lymphedema dle věku vzniku a přidružených symptomů
- Congenital hereditary lymphedema
- Hereditary lymphedema, type I
- Lymphedema-distichiasis
- Lymphedema praecox
- Lymphedema tarda
- Milroy disease
- Nonne-Milroy disease
- Noonan syndrome
- Klippel-Trénaunay-Weber syndrome
- Lymphedema-hypoparathyroid syndrome
- Turner syndrome [3]
Zjednodušeně česky
- Lymfedém, který vzniká bez zjevné příčiny = primární lymfedém
- Do druhého roku života = kongenitální
- Náhodný nebo hereditární (+ OA)
- Ev. součástí jiných vrozených poruch
- Mezi 2.–35. rokem života = lymphoedema praecox
- Po 35. roku života = lymphoedema tardum [8]
Mutace
- Mutations in specific genes important to lymphatic development or function
- Lymphedema
- Chylothorax
- Chylous ascites
- Lymphatic malformations
- Overgrowth syndromes with a lymphatic component
- Germline mutations
- In at least 20 genes that encode proteins acting around VEGFR-3 signaling
- Downstream of other tyrosine kinase receptors
- Effects via the RAS/MAPK and the PI3K/AKT pathways
- More than a quarter of the incidence of primary lymphedema
- Common forms may also result from
- Multigenic effects
- Post-zygotic mutations [5]
Primární kongenitální lymfedém - Hereditary or primary lymphedema - Milroy disease - Nonneho-Milroyův syndrom Hereditární kongenitální lymfedém typu I
Penetrance
- Approximately 85%-90% of individuals who have a mutation in FLT4 develop lower-limb lymphedema by age three years;
- 10%-15% of individuals with an FLT4 mutation are clinically unaffected.
Prevalence
- The prevalence of Milroy disease is not known but it appears to be one of the more common causes of primary lymphedema. [26]
Klinika
Lymfedém
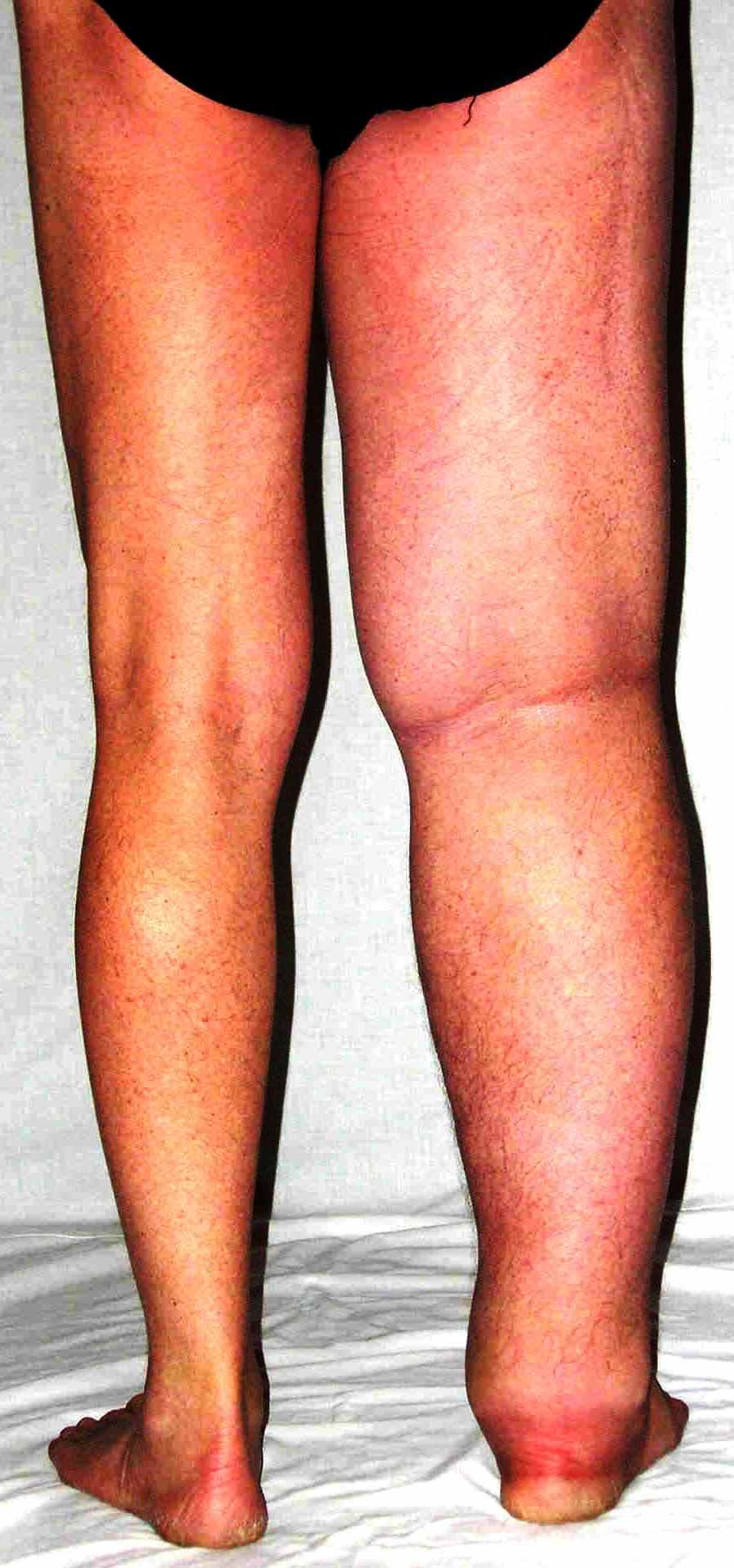
- A subset of patients with congenital lymphedema has a familial sex-linked pattern of inheritance, which is termed Milroy disease. It accounts for 2% of primary lymphedema cases
- Swelling is usually bilateral but can be asymmetric. The degree of edema can progress but in some instances can improve, particularly in early years. [25]
- Developmental disorder of the lymphatic system [3]
- Hypoplasia of peripheral lymphatics, for example, Milroy and Meige disease [8]
- Hypoplastic cutaneous, but not visceral, lymphatic vessels [13]
- The histology of the lymphatic channels often demonstrates an anaplastic pattern without subcutaneous lymphatic trunks but with normal dermal plexus. [12]
- Primary lymphedema has been classified as Milroy disease when present at birth or as Meige disease, which develops predominantly after puberty. Both diseases are characterized by a combination of dilated lymphatic capillaries and interstitial accumulation of lymph fluid leading to lymphedema. [18]
- No uptake of tracer into the lymphatic vessels in the swollen feet of volunteers with Milroy disease, irrespective of whether the volunteer was lying down or sitting. [14]
- Gravity, therefore, does not alter lymphatic function in the same way as shown in lymphoedema distichiasis. [14]
- Lymphatic filling was normal in skin regions with no swelling (forearm). This is a little strange given that the mutation must be present in all cells of the body, but only seems to alter lymphatic function below the knee whilst leaving normal lymphatic function at other sites on the body. [14]
- Postihuje častěji dívky [2]
- Congenital onset (present from birth) [5]
- Ninety per cent of the 71 individuals carrying a VEGFR-3 mutation showed signs of oedema [15]
- Lymphedema of one or both legs and occasionally of the arms, face, or other body parts [19]
- The atypical presentations included pre-natal pleural effusion, spontaneous resorption of lymphedema and elephantiasis.[26]
Celulitida
- Other symptoms and signs included cellulitis (20%) [15]
- Cellulitis, which can damage the lymphatic vessels, occurs in approximately 20% of affected individuals, with infection significantly more likely in males than females. [25]
Varixy
- Large calibre leg veins (23%) [15]
- Prominent veins (23%) [25]
Papillomatosis
- (10%) [15], [25]
Upslanting toenails
- (10%) [15]
- (14%) [25]
Hydrocoele
- The next most common finding after oedema (37%) [15]
- [23]
- Hydrocele (37% of males) [25]
Urethral abnormalities
- In males (4%) [25]
Role VEGFR3 (FLT 4)
- VEGFR-3 is a member of the fms-like tyrosine kinase family [22]
- Specifically binds vascular endothelial growth factor (VEGF)-C and VEGF-D, but not VEGF-A. [22]
- During early embryonic development, VEGFR-3 is expressed both in developing venous and in presumptive lymphatic endothelia. [22]
- In normal adult tissues, VEGFR-3 expression is largely restricted to the lymphatic endothelium. [22]
- VEGFR-3 expression has also been detected on some blood capillaries associated with tumor neovascularization or with wound granulation tissue [22]
- VEGFR-3 alone is not a sufficiently specific marker for lymphatic vessels.[22]
- VEGF-C and VEGF-D were originally cloned as ligands for VEGFR-3
- Are presently the only known ligands for this receptor. [22]
- VEGF-C promotes proliferation, migration, and survival of cultured human LECs
- Transgenic mice overexpressing VEGF-C or VEGF-D in the skin show hyperplasia of cutaneous lymphatic vessels [22]
- VEGF-C null mouse embryos completely lack a lymphatic vasculature and die prenatally of fluid accumulation within the tissues [22]
- The lymphatically committed venous endothelial cells express Prox1 but are unable to migrate out to form the initial lymph sacs [22]
- VEGF-D also stimulates lymphangiogenesis in tissues and tumors [22]
- VEGF-D-deficient mice do not exhibit a lymphatic phenotype [22]
- Probably because VEGF-D is not expressed at the critical sites of lymph sac formation in the embryo [22]
- Inactivation of VEGFR-3 causes cardiovascular failure and embryonic death at E9.5 before the emergence of lymphatic vessels [22]
- VEGFR-3 mutations have been identified in Chy mutant mice that are characterized by cutaneous lymphedema [22]
- VEGF-C and VEGF-D – after enzymatic cleavage – also activate VEGFR-2, which is expressed by LECs [22]
- Skin-specific overexpression of a VEGFR-3-specific mutant of VEGF-C in transgenic mice revealed that activation of VEGFR-3 signal transduction is sufficient to promote lymphangiogenesis [22]
- VEGF-A potently induces proliferation of LECs in vitro
- Injection of adenoviral murine VEGF-A164 resulted in pronounced and persistent in vivo lymphangiogenesis in mouse ear skin [22]
- Mutations in certain critical regions of FLT4, called the tyrosine kinase domains, cause impaired signaling from the VEGFR3 receptor, and result in hypoplasia (underdevelopment) of the lymphatic vessels. [5]
- Encodes a receptor tyrosine kinase specific for lymphatic vessels [4]
- Defective VEGFR3 signaling seems to be the cause of congenital hereditary lymphedema linked to 5q34-q35. [4]
- Mutace VEGFR3 způsobuje dysgenezi lymfatických cév [2]
- Neuropilin (NRP)-2 bound VEGF-C and was expressed in the visceral, but not in the cutaneous, lymphatic endothelia, suggesting that it may participate in the pathogenesis of lymphedema. Virus-mediated VEGF-C gene therapy was able to generate functional lymphatic vessels in the lymphedema mice. [13]
- Lymphedema in patients having ectodermal dysplasia with immunodeficiency may be caused by defective VEGFR-3 signaling via the nuclear factor (NF)-kappa B transcription factor [13]
- VEGFR3 is one of the rare genes expressed almost exclusively in the lymphatic endothelial cells in adults, although it is also needed for proper generation of the embryonic blood vasculature [13]
- Overexpression of the VEGFR-3 ligands VEGF-C and VEGF-D in the skin of transgenic mice induced the formation of a hyperplastic lymphatic vessel network [13]
- Expression of ligand-blocking concentrations of soluble VEGFR-3 in transgenic mice inhibited the development of the lymphatic vasculature in several organs. [13]
- Expecting to be unable to see any lymphatic vessels at all, we were surprised to see some vessels within the skin (possibly a normal level although this analysis is still ongoing). This means that the mutation must be causing a reduction in lymphatic function rather than solely preventing lymphatic vessels from forming. [14]
- VEGFR-3 is the main known trigger on lymphatic cells that when stimulated causes cell multiplication and lymphangiogenesis. [14]
- Overexpression of VEGF-C, a ligand of the VEGF receptors VEGFR-3 and VEGFR-2, in the skin of transgenic mice resulted in lymphatic, but not vascular, endothelial proliferation and vessel enlargement. [15]
- Thus, VEGF-C induces selective hyperplasia of the lymphatic vasculature, which is involved in the draining of interstitial fluid and in immune function, inflammation, and tumor metastasis. [15]
- VEGFR-3 is initially expressed in angioblasts of murine head mesenchyme, dorsal aorta, cardinal vein, and allantois [16]
- VEGFR-3 is expressed both in developing venous and in presumptive lymphatic endothelia, whereas in adult tissues, VEGFR-3 is largely restricted to the lymphatic endothelium [16]
- VEGF-A signalling through VEGFR-2 is important for lymphangiogenesis. [22]
- Indirect effects of VEGF-A, via attraction of inflammatory cells producing VEGF-C and -D might also contribute to VEGF-A's lymphangiogenic activity [22]
- VEGF-C signalling through VEGFR-3 may be enhanced by neuropilin-2, similar to the Nrp1-mediated promotion of VEGF-A signalling to VEGFR-2 [22]
- Ang1 also acts indirectly via the VEGF-C/VEGFR-3 pathway. [22]
- The promigratory effects of HGF are in part mediated by the integrin alpha9, which is specifically expressed by LECs and is required for normal lymphatic function [22]
- Overexpression of HGF in transgenic mice as well as subcutaneous delivery of this growth factor resulted in increased numbers and enlargement of lymphatic vessels. These effects were not inhibited by a VEGFR-3 blocking antibody demonstrating that HGF can directly promote lymphangiogenesis in vivo. [22]
- FGF-2 might also directly act via its receptor FGFR-3, which is upregulated by Prox1 in lymphatic endothelium [22]
- FGF-2 enhances migration and proliferation of primary LECs in vitro, and the promigratory effect could not be abrogated by neutralization of VEGFR-3, raising the possibility that FGF-2 might also function independently of the VEGF-C/VEGFR-3 pathway [22]
- Platelet-derived growth factor-BB and insulin-like growth factors 1 and 2 might also induce lymphangiogenesis in the mouse corneal assay, but their potential effects on skin lymphangiogenesis remain unclear. [22]
- Likely, several lymphangiogenic growth factors work together in a complex way, contributing to the process of lymphatic vessel formation and growth in physiological or pathological conditions [22]
- VEGFR-3 is also expressed in some blood capillaries during tumor neovascularization and in wound granulation tissue, therefore, depending on the tissue and the developmental stage, this molecular marker alone may not precisely discriminate between blood and lymphatic vessels. [16]
- Disruption of Vegfr3 in mice leads to embryonic death at day 9.5 due to defective development of large blood vessels and cardiovascular failure [20]
- During early vascular development, all endothelial cells of the embryonic cardinal vein express two important lymphatic markers, lymphatic vascular endothelial hyaluronan receptor (LYVE-1) and VEGFR-3, and display lymphatic competence. [22]
- Stimulation by a yet unidentified inductive mesenchymal signal leads to induction of the transcription factor Prox1 in a subset of endothelial cells that become committed to the lymphatic lineage. These cells bud off from the vein and migrate into the surrounding tissue to form primitive lymph sacs. During this process, they adopt the expression of additional lymphatic lineage markers. The formation of a mature lymphatic network continues through the first postnatal days. [22]
- VEGF-C156S, a mutant form of VEGF-C that selectively activates VEGFR-3, successfully induced the formation of a functional cutaneous lymphatic vessel network without blood vessel growth or vascular leakiness, side effects observed with VEGF-C gene therapy due to its activation of VEGFR-2 [22]
- Regeneration of a lymphatic network was observed after injection of VEGF-C protein in a surgical lymphedema model in the rabbit ear, indicating the potential use of VEGF-C for the treatment of secondary lymphedema [22]
- Increased levels of VEGF-C and/or VEGF-D promote active tumor lymphangiogenesis and lymphatic tumor spread to regional lymph nodes, and that these effects can be suppressed by blocking VEGFR-3 signalling [22]
- Tumor-derived VEGF-A promotes expansion of the lymphatic network within draining, sentinel lymph nodes, even before these tumors metastasized [22]
- Direct correlation between expression of VEGF-C or VEGF-D by tumor cells and metastatic tumor spread in many human cancers, indicating an important role of lymphangiogenesis also in human tumor progression [22]
- Primary melanomas that later metastasized were characterized by increased lymphangiogenesis – as compared to non-metastatic tumors – and that the degree of tumor lymphangiogenesis can serve as a novel predictor of lymph node metastasis and overall patient survival, independently of tumor thickness. [22]
- Tumor lymphangiogenesis also significantly predicted the presence of sentinel lymph node metastases at the time of surgical excision of the primary melanoma [22]
- Psoriatic skin lesions are characterized by pronounced lymphatic hyperplasia and chronic skin inflammation in mice is also associated with LEC proliferation and lymphatic hyperplasia [22]
- Kidney transplant rejection is frequently accompanied by lymphangiogenesis, and LEC-derived chemokines such as CCL21 might actively promote the inflammatory process [22]
- Acute UVB irradiation of the skin results in hyperpermeable, leaky lymphatic vessels that are functionally impaired [22]
- Blockade of VEGFR-3 resulted in prolonged inflammation and edema after UVB irradiation. [22]
- Lymphatic vessels are not only required to drain inflammation-associated tissue edema, but might also actively participate in the maintenance of chronic inflammatory diseases. [22]
- Blokáda VEGFR-3 vede ke zvýšenému prosakování a zánětu po poškození lymfatik - například vlivem UV záření !!!
- Tedy zhoršená regenerace funkce lymfatických cév. Tedy obdobné se děje u mutací ? Mutace - poškození - delší zánět s prosakem, následná lymfoproliferace a stále dokola, až je výsledkem hyperplazie lymf. systému s mnoha lymf. cévami, které prosakují a špatně fungují - dilatují a jen velmi pomalu obnovoují svoji funkci...a jednou, když se poškodí příliš moc lymfatických cév naráz vznikne až lymfedem....typicky po poštípání hmyzem, očkování atd...ale i samovolně z nezn. příčin....
- ...a tedy pak by bylo možné vysvětlit i ony raritní případy, kdy ojediněle může dojít k resorpci a regresi primárního lymfedému ? Jako že dlouho nevznikne žádná další vyvolávající příčina, která by opět zhoršila funkci a lymfatické cévy se zvládnou po nějaké době přecijenom ještě nějak regenerovat?
- Je možné v těle vyprovokovat produkci nějaké obdobné látky jako VEGFR-3, která by nahradila onu zmutovanou?
- Treatment of lymph node–excised mice with adenovirally delivered vascular endothelial growth factor-C (VEGF-C) or VEGF-D induced robust growth of the lymphatic capillaries, which gradually underwent intrinsic remodeling, differentiation and maturation into functional collecting lymphatic vessels, including the formation of uniform endothelial cell-cell junctions and intraluminal valves. The vessels also reacquired pericyte contacts, which downregulated lymphatic capillary markers during vessel maturation. Growth factor therapy improved the outcome of lymph node transplantation, including functional reconstitution of the immunological barrier against tumor metastasis. --Growth factor–induced maturation of lymphatic vessels is possible in adult mice and provide a basis for future therapy of lymphedema. [27]
- Soluble form of VEGFR-3 is a potent inhibitor of VEGF-C/VEGF-D signaling, and when expressed in the skin of transgenic mice, it inhibits fetal lymphangiogenesis and induces a regression of already formed lymphatic vessels, though the blood vasculature remains normal. [28]
- Transgenic mice develop a lymphedema-like phenotype characterized by swelling of feet, edema and dermal fibrosis. [28]
- They survive the neonatal period in spite of a virtually complete lack of lymphatic vessels in several tissues, and later show regeneration of the lymphatic vasculature, indicating that induction of lymphatic regeneration may also be possible in humans. [28]
- The gene encoding the receptor tyrosine kinase VEGFR-3 is one of the genes upregulated by Prox-1 in the lymphatic endothelial cells. VEGFR-3 is required for remodeling of the blood vascular network at midgestation, but becomes downregulated in the blood vessel endothelia after the emergence of the lymphatic vessels. [28]
- VEGFR-3 activation by its ligands VEGF-C and VEGF-D leads to proliferation, migration and survival of cultured human adult microvascular lymphatic endothelial cells. [28]
- VEGFR-3 activation also induces lymphangiogenesis in adult tissues [28]
- Missense mutations in the gene encoding VEGFR-3 lead to insufficient signaling through this receptor and hypoplasia of the cutaneous lymphatic network in a subset of families suffering from congenital human lymphedema [28]
- The two known VEGFR-3 ligands, VEGF-C and VEGF-D, belong to the larger VEGF family of growth factors that also includes VEGF, placenta growth factor and VEGF-B. [28]
- In addition to activating VEGFR-3, VEGF-C and VEGF-D also activate VEGFR-2, which is expressed in both blood and lymphatic vessel endothelia. [28]
- Proteolytic cleavage is an important regulator of the receptor binding and thus the biological activity of VEGF-C and VEGF-D. [28]
- Partially processed forms of VEGF-C and VEGF-D activate VEGFR-3, whereas the fully processed short forms are also potent stimulators of VEGFR-2. [28]
Mutace VEGFR3 (FLT 4)
- Autosomal dominant trait with incomplete penetrance [3]
- Variable expression, and variable age at onset [13]
- Sequence analysis. The mutation detection frequency using sequence analysis is currently unknown as limited data are available. [24]
- Up to 75% can be expected in those clearly affected and with a positive family history. [24]
- If those with typical Milroy features but without a family history are included the mutation pick-up rate is around 68%. [24]
- Individuals with an FLT4 mutation have a 1 in 2 (50%) chance of passing that mutation on to each child, theoretically leading to lymphedema. However, only about 85-90% of those who inherit an FLT4 mutation will actually develop lymphedema, a phenomenon called incomplete penetrance. [5]
- Exhibits variable expression, which means that there can be differing levels of severity of the lymphedema in different family members [5]
- Most mutations in the FLT4 gene have been found in 8-10 of these exons, and therefore, sequencing of this gene can initially be targeted to these areas, reducing the cost of screening to approximately $500* [5]
- Mutace receptoru pro endoteliální růstový faktor v cévách [1] - vascular endothelial growth factor C receptor VEGFR3 was mapped to the linked region, and partial sequence analysis identified a G-->A transition at nucleotide position 3360 of the FLT4 cDNA, predicting a leucine for proline substitution at residue 1126 of the mature receptor in one nuclear family [4]
- Mapped to the telomeric part of chromosome 5q, in the region 5q34-q35 [4]
- Linkage of the disease with markers in 5q34-q35, including a VEGFR3 intragenic polymorphism [4]
- A-->G transition that cosegregates with the disease, corresponding to a histidine-to-arginine substitution in the catalytic loop of the protein. [4]
- Chy mice with viral VEGF-C gene therapy, we were able to induce the growth of functional lymphatic vessels in their skin. Milroy's disease would thus provide one example of a human hereditary disease where gene therapy seems feasible, and it could provide a paradigm for other diseases associated with mutant receptors. [13]
- Because of possible complications due to tissue edema or accelerated tumor angiogenesis, the VEGFR-3-specific growth factor VEGF-C156S could thus be a more attractive choice for therapeutic applications. [13]
- VEGF-C administration alone or in combination with other lymphangiogenic factors could be a powerful tool in the therapy of various forms of human lymphedema [13]
- Phenotype and markers in the 5q34-q35 region, and describe an H1035R substitution in a highly conserved residue in the catalytic loop of the VEGFR3 receptor. This amino acid substitution causes loss of VEGFR3 tyrosine kinase activity. Thus, defective VEGFR3 signaling is responsible for 5q34-q35–linked lymphedema. [20]
- The H1035R mutation is located in the catalytic domain of VEGFR3, not in a region of tyrosyl autophosphorylation sites. Thus, the loss of autophosphorylation capacity by H1035R indicates loss of tyrosine kinase activity, rather than lack of kinase site recognition, and lymphedema is caused by lack of sufficient VEGFR3 signaling. [20]
Vyšetření
- Ultrasonography in the third trimester of pregnancy may detect swelling of the dorsum of the feet and more extensive edematous states in an affected fetus. [25]
- Prenatal testing is possible for pregnancies at increased risk if the disease-causing mutation in the family is known; however, it is rarely requested. [25]
Literatura:
[1] www.zdn.cz/clanek/postgradualni-medicina/lymfedem-156496
[2] www.praktickagynekologie.cz/pdf/pg_04_06_03.pdf
[3] www.ncbi.nlm.nih.gov/pubmed/9817924
[4] www.ncbi.nlm.nih.gov/pubmed/10856194
[5] www.lymphnotes.com/article.php/id/488/
[6] jmg.bmj.com/content/37/9/725.extract
[7] www.cell.com/AJHG/abstract/S0002-9297%2810%2900213-2
[8] www.nature.com/ejhg/journal/v16/n3/full/5201982a.html
[9] webeye.ophth.uiowa.edu/eyeforum/cases/84-Distichiasis-Extra-Eyelashes.htm
[10] emedicine.medscape.com/article/1212908-overview
[11] jmg.bmj.com/content/39/7/478.abstract
[12] emedicine.medscape.com/article/191350-overview
[13] www.pnas.org/content/98/22/12677.full
[14] www.lymphoedema.org/Menu3/5Articles%20by%20healthcare%20professionals.asp
[15] jmg.bmj.com/content/42/2/98.abstract
[16] genesdev.cshlp.org/content/16/7/773.full
[17] humrep.oxfordjournals.org/content/14/3/823.full
[18] genesdev.cshlp.org/content/16/7/773.full
[19] radiographics.rsna.org/content/20/6/1697.full
[20] www.ncbi.nlm.nih.gov/pmc/articles/PMC1287178/?tool=pubmed
[21] www.ncbi.nlm.nih.gov/omim/153400
[22] www.nature.com/jid/journal/v126/n10/full/5700464a.html
[23] www.uklymph.com/latest_news/ln_inheritance_of_primary_lymphedema.php
[24] www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=milroy
[25] www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=milroy
[26] onlinelibrary.wiley.com/doi/10.1111/j.1399-0004.2006.00687.x/full
[27] www.nature.com/nm/journal/v13/n12/full/nm1689.html
[28] www.nature.com/nm/journal/v7/n2/full/nm0201_199.html
Distichiasis-lymphedema syndrome
- Rare autosomal dominant
- Multisystem disorder
- Swelling due to fluid accumulation
- Usually around puberty
- Extra eyelashes along the posterior border of the lid margins (distichiasis)
- May range from a few extra lashes to a full set of extra eyelashes
- Swelling most often affects the legs
- Cleft palate
- Droopy eyelids (ptosis)
- Abnormalities of the curved transparent outer layer of fibrous tissue covering the eyeball (cornea)
- Cysts on the spinal cord
- Abnormal sensitivity to light (photophobia)
- Cardiac (heart) defects
- Caused by mutations of the FOXC2 gene [3]
Hereditary lymphedema type IA - Nonne-Milroy’s disease
Klinika
- Swelling (edema) present at or shortly after birth (congenital)
- Rare cases, edema may develop later in life
- The legs are most often affected
- The extent and location of edema varies greatly
- Genitals may also be affected
- Additional complications sometimes
- Upslanting toenails
- Small warty growths on the affected areas (papillomatosis)
- Abnormally large or prominent leg veins
- In males
- Urethral abnormalities
- Fluid-filled sac along the spermatic cord of the scrotum (hydrocele)
Genetika
- Mutation in the FLT4 gene
- Encodes VEGFR-3 gene
- Located on the 5q35.3 [3]
Hereditary lymphedema type II - Meige disease - lymphedema praecox
Klinika
- Around puberty or shortly thereafter in most individuals - peripubertal
- Most common type of primary lymphedema
- Cca 80 percent of cases [3]
- Lymphedema of the legs
- And other areas of the body may be affected:
- The arms
- Face
- Larynx
- Yellow nails in some people [3]
Genetika
- Mutations of the ‘forkhead’ family transcription factor (FOXC2) gene
- Located on the long arm (q) of chromosome 16 (16q24.3) [3]
- Different mutations of the same gene (FOXC2) disorders include:
- Yellow nail syndrome
- Distichiasis-lymphedema syndrome
- Lymphedema-ptosis syndrome [3]
Lymphedema-ptosis syndrome
- Extremely rare genetic disorder
- Swelling because of fluid accumulation
- Most often affects the legs
- Droopy eyelids (ptosis)
- Usually occurs at or shortly after puberty
- Mutations of the FOXC2 gene
- Autosomal dominant [3]
Lymphedema tarda - late on-set lymphedema
- Primary lymphedema
- After the age of 35
- Legs are most often affected
- Arms and other areas may be affected as well [3]
Meige disease - Lymphedema tarda/praecox - Lymphedema hereditary type 2
Klinika
- Clinically apparent during puberty or later onset [8],[6] 35 years or older [12]
- Edema, particularly severe below the waist, develops about the time of puberty. [14]
- Involvement of the upper limbs (as well as the lower limbs), face, and larynx [14]
- In one, a persistent pleural effusion [14]
- Scintilymphangiography indicated paucity or absence of lymph nodes in the axillae and above the inguinal ligaments [14]
- Chronic facial swelling - characteristic appearance of affected members including puffiness, shiny skin, deep creases, and, in some, excessive wrinkling [14]
- Association of late-onset lymphedema with deafness [14]
- With primary pulmonary hypertension [14]
- Cerebrovascular malformations [14]
- Association of cleft palate [14]
- Thrombophlebitis, episodic waxing and waning of lymphedema, lymphangiosarcoma on the inner right thigh and died of metastases some months later. [14]
- Andersson et al. (1995) raised the question of whether a genetic predisposition to malignancy combined with the lymphedema was etiologically significant [14]
- There seemed to be an unusually high frequency of cancer (uterine, colon, lung, prostate, breast, and bone) in the proband's family. [14]
- The most common form of the primary lymphedema [8]
- Oedema clinically indistinguishable from that found in the lymphoedema–distichiasis syndrome [8]
- Hypoplasia of peripheral lymphatics like Milroy disease [8]
- ?
- Histologically - likely a hyperplastic pattern, with tortuous lymphatics increased in caliber and number. They often display absent or incompetent valves. [12]
- ?
- Rarest form of primary lymphedema [12]
- Accounts for only 10% of cases [12]
- Presents with pubertal-onset lymphedema. No other features appear to be associated. Women are more commonly affected than men. No genes have been identified as yet. Inheritance appears to be autosomal dominant with reduced penetrance.[15]
Mutace FOXC2
- Perhaps the most well known of genes responsible for hereditary lymphedema is the FOXC2 gene. Identified by University of Michigan scientists and collaborators from the University of Arizona, FOXC2 is the genetic cause of Lymphedema Praecox Meige Syndrome (Hereditary Lymphedema II). [13]
- Hereditary lymphedema type II is caused by mutation in the forkhead family transcription factor gene MFH1 . Allelic disorders with overlapping features include the lymphedema-distichiasis syndrome, lymphedema and ptosis, and lymphedema and yellow nail syndrome. [14]
- Phenotypic classification of autosomal dominant lymphedema does not reflect the underlying genetic causation of these disorders [14]
Literatura:
[6] jmg.bmj.com/content/37/9/725.extract
[8] www.nature.com/ejhg/journal/v16/n3/full/5201982a.html
[12] emedicine.medscape.com/article/191350-overview
[13] www.lymphedemapeople.com/wiki/doku.php?id=lymphedema_gene_foxc2
[14] www.lymphedemapeople.com/wiki/doku.php?id=lymphedema_gene_foxc2
[15] www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=milroy
Yellow nail syndrome
- Rare disorder
- Yellow, thickened, and curved nails
- With almost complete stoppage of nail growth
- Loss of the strip of hardened skin at the base and sides of a fingernail (cuticles) may also occur
- Separation of the nails from the nail bed (onycholysis)
- May cause the nails to fall out
- Usually associated with :
- Presence of fluid in the lungs (plural effusion)
- Swelling of the arms and legs (lymphedema)
- Other respiratory problems may occur
- Chronic inflammation of the bronchi and bronchioles (bronchiectasis)
- Chronic bronchitis
- Inflammation of the membranes in sinus cavities (sinusitis)
- Lymphedema usually occurs around puberty
- Mutations to the FOXC2 gene autosomal dominant [3]
CLOVES
- Congenital lipomatous overgrowth,
- Vascular malformations,
- Epidermal nevi,
- Skeletal/spinal abnormalities syndromes [56]
Lymphedema-Distichiasis (LD) Syndrome - Like Milroy’s Disease - LD Syndrome - Lymphoedema with ptosis
Klinika
Lymphedema
- Hyperplasia with reflux [8]
- Lower extremities
- Asymmetric
- Variable age of onset
- Usually develops around puberty in both males and females, or during pregnancy in females [6]
- Males develop edema at an earlier age and have more problems with cellulitis [27]
- Lymphatics in the feet could function normally when at heart level (lying down), but did not work as well when below heart level (sitting or standing) [14]
- Bilateral hypoplasia lymphedema is an uncommon form of lymphedema in which lymphography shows abundant, dilated lymphatics occurring in both lower limbs, and the thoracic duct is either absent, obstructed, or deformed. [21]
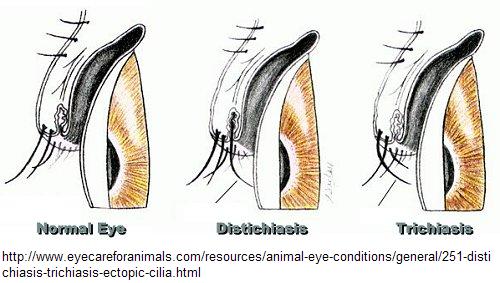
Distichiasis (double rows of eyelashes)
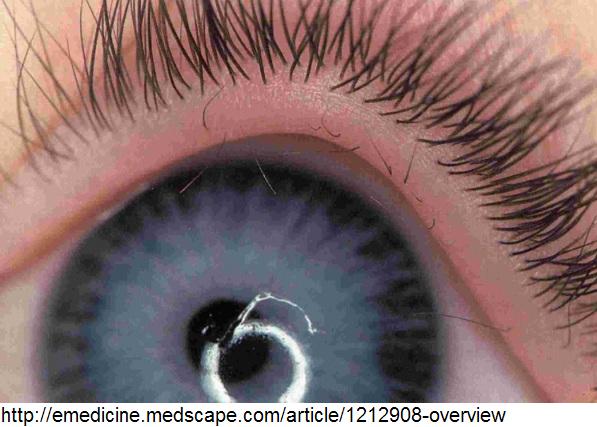
- Secondary eyelashes originating from the site of meibomian glands openings [6], in 94% of those with mutations [8]
- 94% penetrant, similar to the penetrance level of lymphoedema [8]
- Asymptomatic distichiasis appears to be more common than might be expected.After careful examination, advice on which genes to screen can be given. [8]
- Longstanding irritation of both eyes - chybná terpaie pro "blepharitis" and allergic conjunctivitis. Despite using olopatadine (Patanol) drops and lid hygiene, his symptoms persistented. [9]
- Acquired distichiasis is less common, and is seen in the setting of chronic inflammatory conditions of the eyelid such as blepharitis, ocular cicatricial pemphigoid, Steven Johnson syndrome, and following chemical injury. Acquired distichasis tends to be more symptomatic than its congential counterpart. [9]
- Distichiasis most commonly affects the dog, especially the American and English Cocker Spaniels, Miniature and Toy Poodles, English Bulldogs, and Golden Retreivers. Cats are usually not affected because they lack true cilia. [11]
- About 75% of affected individuals have ocular findings resulting from the aberrant eyelases including corneal irritation, recurrent conjunctivitis, and photophobia [27]
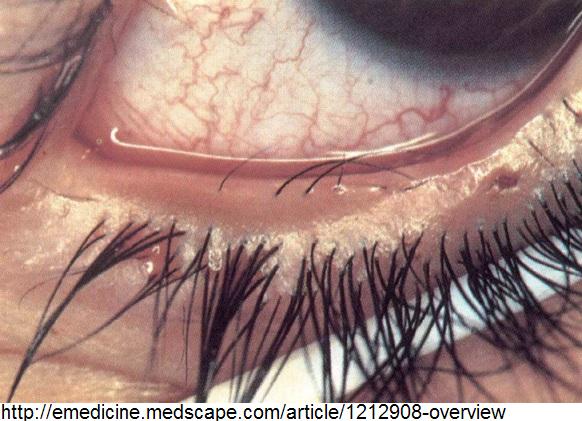
Early varicose veins
- 49% procent případů [8]
- Varicose veins, present in 49%, were notable for early onset and increased prevalence compared to the general population [25]
- Volunteers (few in number) who had mutations in the gene but no swelling (carriers) still had venous abnormalities but normal lymphatic function. This suggests that the mutations firstly affect the venous system over the lymphatic system, but that a dysfunctional lymphatic system is required to ensure the development of swelling [14]
- Venous reflux in 100% of those individuals with a FOXC2 mutation, regardless of their lymphoedema status [8]
- Possible developmental role for FOXC2 in both venous and lymphatic systems. This is the first gene that has been implicated in the aetiology of varicose veins [11]
Ptosis
- Droopy eyelids - 31% pacientů s mutací [8]
Cleft palate
- 4% pac. s mutací [8]
- About 4% of individuals have cleft palate with or without Pierre-Robin sequence. [28]
Cardiac abnormalities
- Congenital heart disease occurs in 7% of individuals with lymphedema-distichiasis syndrome. Structural abnormalities include ventricular septal defect, atrial septal defect, patent ductus arteriosis, and tetralogy of Fallot. Cardiac arrhythmia, most commonly sinus bradycardia, may also occur. [28]
- Congenital heart disease (8%) [28]
Spinal extradural cysts
- Spinal extradural cysts are often arachnoid diverticula. [21]
- Scoliosis, spinal extradural cysts, neck webbing, uterine and renal anomalies, strabismus, and synophrys. Neonatal chylothorax has been reported in one case only in association with congenital heart disease [28]
- Association with yellow nails, but discolored nails are often a feature of chronic lymphedema regardless of cause. [28]
Photophobia
Renal abnormalities
- Including nephritis, duplex kidney, and recurrent infections. [21]
Pulmonary lymphangiectasia
- Bilateral pleural effusion/chylothorax and hydrops - died neonatally [26]
- Prenatal intrauterine hemithoracic drainage [26]
- Congenital pulmonary lymphangiectasia (CPL) [26]
Hydrops
- Subcutaneous edema and effusions in two or more body cavities [26]
- May be due to a variety of factors [26]
- Supports the notion that chylothorax and hydrops may be caused by structural lesions of lymph channels [26]
- Autosomal recessive inheritance, with intrafamilial variability of a lymphatic disorder on a genetic basis [26]
- Mutations in vascular endothelial growth factor receptor-3 (VEGFR 3) in families with Milroy disease, mutations of FOXC2 in the lymphedema-distichiasis syndrome, and fatal chylothorax in alpha9-deficient mice are potential candidate genes [26]
Without other syndromologie
- About 25% of individuals are asymptomatic
Blepharocheilodontic syndrome
- The association of lagophthalmos (inability to fully close eyes), cleft lip and palate, atrial septal defect, and oligodontia. Distichiasis is a feature; lymphedema is not observed. [28]
Další
- Lymphoedema and distichiasis - 1 patient born at 31 weeks gestation, had a patent ductus arteriosus and pyloric stenosis and her father had a deep vein thrombosis [8]
Funkce FOXC2
- FORKHEAD BOX C2; FOXC2 - The 'forkhead' (or winged helix) gene family, originally identified in Drosophila, encodes transcription factors with a conserved 100-amino acid DNA binding motif. [26]
- Alternative titles; symbols FORKHEAD, DROSOPHILA, HOMOLOG-LIKE 14; FKHL14 MESENCHYME FORKHEAD 1; MFH1 LYMPHEDEMA-DISTICHIASIS SYNDROME WITH RENAL DISEASE AND DIABETES MELLITUS, INCLUDED [26]
- Gene map locus 16q24.3 [26]
- Broad phenotypic heterogeneity was observed within these families.
- 4 overlapped phenotypically defined lymphedema syndromes:
- Meige lymphedema
- Lymphedema-distichiasis syndrome
- Lymphedema and ptosis
- Yellow nail syndrome
- But not Milroy disease [26]
- Phenotypic classification of autosomal dominant lymphedema does not appear to reflect the underlying genetic causation of these disorders. [26]
- FOXC2 codes for a protein (transkripční faktor) that regulates the activity of several other genes that are involved in the development of several organs and systems in the fetus [5]
- FOXC2 as a key regulator of adipocyte metabolism. [26]
- In mice overexpressing Foxc2 in white adipose tissue (WAT) and brown adipose tissue (BAT), the intraabdominal WAT depot was reduced and had acquired a brown fat-like histology, whereas interscapular BAT was hypertrophic. [26]
- Increased Foxc2 expression had a pleiotropic effect on gene expression in BAT and WAT. There was an induction of the BAT-specific gene Ucp1 in the intraabdominal WAT depot. The authors also demonstrated a change in steady-state levels of several WAT- and BAT-derived mRNAs that encode genes of importance for adipocyte insulin action, differentiation, metabolism, sensitivity to adrenergic stimuli, and intracellular signaling. The nature of these Foxc2-generated responses was consistent with protection against obesity and related symptoms, such as diet-induced insulin resistance. [26]
- Zvyšuje aktivitu spalování v tukové tkáni?
- Porucha u lymfedému s X foxc2 = tendence tukové tkáně méně spalovat energii - snažší usazení se?
- In wildtype mice, Foxc2 mRNA levels were upregulated by high-fat diet,whereas mice with targeted disruption of 1 Foxc2 allele had a decreased interscapular BAT cell mass. [26]
- X foxc2 vede k hyperplazii lymfatik s dysfunkcí, zlepší vysokotuková dieta stav?
- FOXC2 affects adipocyte metabolism by increasing the sensitivity of the beta-adrenergic cAMP protein kinase A (PKA) signaling pathway through alteration of adipocyte PKA holoenzyme composition.
- Increased FOXC2 levels induced by high fat diet seem to counteract most of the symptoms associated with obesity, including hypertriglyceridemia and diet-induced insulin resistance, and a likely consequence would be protection against type II diabetes. [26]
- Mají pacienti s mutací FOXC2 vyšší riziko vzniku DM2 ?
- A family of German-Irish descent with lymphedema-distichiasis syndrome in 6 members over 3 generations. In addition to LD, 4 had renal disease and 3 had type II diabetes. [26]
- All affected members were found to have a frameshift mutation in the FOXC2 gene [26]
- All affected and unaffected members of the family were homozygous for the T allele of the 512C-T polymorphism in the 5-prime UTR of the FOXC2 gene. This polymorphism had been found to be associated with insulin sensitivity in Swedish persons but not in Japanese or Pima Indians [26]
- Phenotype of LD, renal disease, and diabetes might be the result of a combination of the mutation and the polymorphism. [26]
- There was no relation between BMI and FOXC2 mRNA in either adipose or muscle. There was a strong inverse relation between adipose FOXC2 mRNA and insulin sensitivity, using the frequently sampled IV glucose tolerance test (r=-0.78, P<0.001). When compared to the insulin sensitive subjects, the insulin resistant subjects had 3-fold higher levels of adipose FOXC2 mRNA. In contrast, muscle FOXC2 mRNA expression was no different between insulin resistant and insulin sensitive subjects. [26]
- Most mutations in FOXC2 result in a protein product that is unable to effectively regulate the activity of certain genes involved in the development of the lymphatic system and other areas affected by the LD Syndrome. [5]
- Je jejich tuková tkáň méně citlivá vůči adrenalinu a noradrenalinu?
- U sekundárního lymfedému - regenerace lymfatik - je tam vyšší (nebo nižší ?) exprese FOX2 a tím (?) vyšší citlivost tukové tkáně k inzulínu a vyšší tendence růstu tukové tkáně?
- Remodelling and maturation of the lymphatic system (we believe specifically the valves) [14]
- Involved in specifying mesenchymal cell fate during embryogenesis, was associated with metastatic capabilities of cancer cells. [26]
- Mají pac. s mutacemi fox2 méně agresivní nádory?
- Foxc2 expression was required for murine mammary carcinoma cells to metastasize to lung, and overexpression of Foxc2 enhanced their metastatic potential. FOXC2 was induced in human and mouse cells undergoing epithelial-mesenchymal transitions (EMTs) triggered by a number of signals. [26]
- Mají méně metastáz v případě karcinomů?
- Netrpí vůbec invazivním duktálním Ca prsu?
- FOXC2 specifically promoted mesenchymal differentiation during an EMT, suggesting that FOXC2 orchestrates the mesenchymal component of the EMT program. FOXC2 was significantly overexpressed in the highly aggressive basal-like subtype of human invasive ductal breast cancer. [26]
- U chronických zánětů v lymfedémech je častější vznik maligních mezenchymových tumorů - lymfangiosarkomů - souvisí to se zvýšenou snahou o regeneraci lymfatik s hyperexpresí nemutovaného FOX2, které pak pomáhá v progresi Ca procesu?
Mutace FOXC2
LYMPHEDEMA-DISTICHIASIS SYNDROME FOXC2, TYR99TER
- C-G change at nucleotide 297
- Tyr99-to-ter (Y99X) substitution in the FOXC2 gene
- Hydrops fetalis 2x, karyotype was 46,XX.
- The father - hereditary lymphedema-distichiasis
- 2 sons had distichiasis [26]
- 4-nucleotide (GGCC) duplication at position 1093 of the coding region of the FOXC2 gene
- 98 novel amino acids before truncating the protein, lay in the carboxy-terminal region after the forkhead domain
- Lymphedema + distichiasis and cystic hygroma, arachnoid cysts, and cleft palate. [26]
- 11-bp deletion involving nucleotides 290-300
- Creation of 361 novel amino acids beginning at codon 96. [26]
- Deletion of 1331A, disrupting codon 443, producing a frameshift, and adding 27 novel amino acids. [26]
- Insertion of a T after nucleotide 209, causing disruption of codon 70 and a frameshift with addition of 391 novel amino acids. [26]
- Dinucleotide insertion of CT after nucleotide 201, disrupting codon 67 and causing a frameshift [26]
- Production of 4 novel amino acids [26]
YELLOW NAIL SYNDROME, INCLUDED
- Single base insertion of C after nucleotide 589
- Frameshift with premature termination at amino acid 463 [26]
- Age of onset was after puberty [26]
- 1 affected family member had a cleft palate. [26]
- Three of 7 affected family members also exhibited ptosis [26]
- Single base deletion of 505A [26]
- Frameshift with premature termination at amino acid 202 [26]
- Age of onset of lymphedema ranged from 8 to 13 years [26]
- Autosomal dominant segregation of upper- and lower-eyelid distichiasis in 7 relatives over 3 generations [26]
- Below-knee lymphedema of pubertal onset in 3
- Two children had cleft palate in addition to distichiasis, but without the previously reported association of Pierre Robin sequence [26]
- Other ophthalmologic anomalies included divergent strabismus and early-onset myopia [26]
- No family member had pterygium colli, congenital heart disease, or facial dysmorphism, the disorder was linked to markers on chromosome 16q24.3 and was thus proposed to be allelic to lymphedema-distichiasis syndrome [26]
- Out-of-frame deletion of the FOXC2 gene, 914-921del, segregating with the syndrome [26]
- 6 affected members spanning 3 generations of a German-Irish family [26]
- With lymphedema-distichiasis syndrome [26]
- 1-bp insertion (1006insA) in the FOXC2 gene [26]
- Frameshift mutation that predicted a premature stop at codon 462 [26]
- Renal disease [26]
- 3 had type II diabetes mellitus [26]
- 5-prime untranslated region for the 512C-T polymorphism showed the homozygous T allele in all family members tested [26]
- Earliest affected member of the family was 73 years old at the time of report and was on chronic renal dialysis [26]
- One of her sons, aged 45 years, had developed proteinuria at age 32 years - chronic sclerosing glomerulopathy and chronic tubulointerstitial nephritis [26]
- One member of the family renal transplantation and, shortly thereafter, pancreatic transplantation, both with excellent results. She was 36 years old at the time of report and had distichiasis but no lymphedema [26]
- The novel phenotype of LD with renal disease and type II diabetes might be the result of a combination of the 1-bp coding region insertion and homozygosity for the T allele of the upstream UTR 512C-T polymorphism www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=602402&rn=1 [26]
- Has dominant inheritance with variable expression and incomplete penetrance [5]
- Majority of reported FOXC2 mutations are small insertions/deletions, a nonsense mutation was identified in a large family with LD [8]
- Only one exon, and is therefore less costly than FLT4 for genetic testing (approximately $200-400*)[5]
- In one family in 8 relatives over three generations [8]
- Frameshift that predicts a premature stop at nucleotide 599 and truncating the normal protein by 38% [8]
- Lymphoedema without distichiasis, all but one of his affected relatives who carried the FOXC2 mutation did have accessory eyelashes originating from their meibomian glands [8]
- Only lymphoedema with distichiasis is caused by FOXC2 mutations [8]
- Careful phenotyping for distichiasis - may be difficult to confirm unless several family members are examined, and cannot ever be assumed to be absent from self-report [8]
- In a family with lymphoedema–distichiasis syndrome [8]
- Males had an earlier onset of lymphoedema and a significantly increased risk of complications. Lymphatic imaging confirmed the earlier suggestion that LD is associated with a normal or increased number of lymphatic vessels rather than the hypoplasia or aplasia seen in other forms of primary lymphoedema. [11]
- (OMIM 153000) [8]
The most well known of genes responsible for hereditary lymphedema is the FOXC2 gene.
- Identified by University of Michigan scientists and collaborators from the University of Arizona, FOXC2 is the genetic cause of Lymphedema Praecox Meige Syndrome (Hereditary Lymphedema II)and as a causative factor in lymphedema-distichiasis syndrome, lymphedema ptosis and lymphedema yellow nail syndrome. [26]
Prenatal Testing
Molecular genetic testing.
- Prenatal diagnosis for pregnancies at increased risk is possible in the US (availability may vary by country) by analysis of DNA extracted from fetal cells obtained by chorionic villus sampling (CVS) at about ten to 12 weeks' gestation or amniocentesis usually performed at about 15-18 weeks' gestation. [28]
- The disease-causing mutation of an affected family member must be identified before prenatal testing can be performed. [28]
- Although available, prenatal diagnosis for lymphedema-distichiasis syndrome is rarely requested. [28]
- Preimplantation genetic diagnosis (PGD) may be available for families in which the disease-causing mutation has been identified in an affected family member. For laboratories offering PGD, see graphic element. [28]
Ultrasonography.
- Fetal echocardiography at 16 to 20 weeks' gestation is recommended because of the increased risk for congenital heart disease. [28]
- Note: Gestational age is expressed as menstrual weeks calculated either from the first day of the last normal menstrual period or by ultrasound measurements. [28]
Literatura:
[1] www.zdn.cz/clanek/postgradualni-medicina/lymfedem-156496
[2] www.praktickagynekologie.cz/pdf/pg_04_06_03.pdf
[3] www.ncbi.nlm.nih.gov/pubmed/9817924
[4] www.ncbi.nlm.nih.gov/pubmed/10856194
[5] www.lymphnotes.com/article.php/id/488/
[6] jmg.bmj.com/content/37/9/725.extract
[7] www.cell.com/AJHG/abstract/S0002-9297%2810%2900213-2
[8] www.nature.com/ejhg/journal/v16/n3/full/5201982a.html
[9] webeye.ophth.uiowa.edu/eyeforum/cases/84-Distichiasis-Extra-Eyelashes.htm
[10] emedicine.medscape.com/article/1212908-overview
[11] jmg.bmj.com/content/39/7/478.abstract
[12] emedicine.medscape.com/article/191350-overview
[13] www.pnas.org/content/98/22/12677.full
[14] www.lymphoedema.org/Menu3/5Articles%20by%20healthcare%20professionals.asp
[15] jmg.bmj.com/content/42/2/98.abstract
[16] genesdev.cshlp.org/content/16/7/773.full
[17] humrep.oxfordjournals.org/content/14/3/823.full
[18] genesdev.cshlp.org/content/16/7/773.full
[19] radiographics.rsna.org/content/20/6/1697.full
[20] www.ncbi.nlm.nih.gov/pmc/articles/PMC1287178/?tool=pubmed
[21] www.ncbi.nlm.nih.gov/omim/153400
[22] www.nature.com/jid/journal/v126/n10/full/5700464a.html
[23] www.uklymph.com/latest_news/ln_inheritance_of_primary_lymphedema.php
[24] www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=milroy
[25] www.lymphedemapeople.com/thesite/lymphedema_distichiasis.htm
[26] www.lymphedemapeople.com/wiki/doku.php?id=lymphedema_gene_foxc2
[27] www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=milroy
[28] www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=lds
Klippel-Trenaunay disorder
- Often associated with venous (Klippel-Trenaunay-Servelle syndrome) and lymphatic abnormalities
- Occasionally coexists with arterial disturbances (Klippel-Trenaunay-Weber syndrome), and seems to arise as a somatic mutation in utero.
Lymphedema-Hypotrichosis-Telangiectasia Syndrome
Klinika
- Lower extremities and occasionally the eyelids and scrotum [5]
- Variable onset, but usually develops by puberty if not present at birth.[5]
- Hypotrichosis (sparse hair) is usually apparent by age 1 and can manifest with absent eyebrows and eyelashes [5]
- Telangiectasias, red spots caused by dilated blood vessels, can be found on the legs, feet, hands, scrotum, and scalp [5]
- Association of childhood-onset lymphedema in the lower limbs, loss of hair, and telangiectasia, particularly on the palms. Inheritance is either autosomal dominant or autosomal recessive. Mutations in SOX18 are causative [15]
Mutace SOX18
- Both dominant and recessive inheritance patterns [5]
- Because the gene is small, with only 2 exons, it is approximately $500* to screen [5]
- Very rare condition [8]
Role SOX18
- A gene involved in the proliferation and migration of cells to the lymph sacs from the veins [14]
- Early malformation in lymphatic development [14]
Literatura:
[5] www.lymphnotes.com/article.php/id/488/
[8] www.nature.com/ejhg/journal/v16/n3/full/5201982a.html
[14] www.lymphoedema.org/Menu3/5Articles%20by%20healthcare%20professionals.asp
[15] www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=milroy
Yellow nail syndrome - lymphoedema and yellow nail syndrome -YNS
Klinika
Nehty
- Frequently show ridging due to interrupted growth. Onycholysis can occur in one or more nails. [14]
- Discoloured, slow growing and excessively curved nails (OMIM 153300), but it remains translucent and smooth whereas in lymphoedema not associated with YNS, the nails become thickened, rough and opaque. [8]

- Yellow nails can be found in MD, MGD and LD, and ptosis occurs in LD [8]
- Yellow-green discoloration, transverse and longitudinal overcurvature, onycholysis, shedding, cross-ridging, and loss of lunulae and cuticles [14]
- Age at onset in the majority of the patients was after 50 years [14]
- Four of the 11 patients had complete recovery of their nails over a mean period of 4 to 5 years [14]
- Sporadic acquired condition rather than a dominantly inherited condition, as currently classified. [14]
Plicní
- Associated features such as chronic sinusitis, bronchiectasis or pleural effusion are essential for a diagnosis of YNS [8]
- Nonimmune hydrops and a recurrent left chylothorax to a mother with the yellow nail syndrome. The nonimmune hydrops in this case was diagnosed on a 29-week ultrasound examination. [14]
- Bilateral chylothorax [14]
Obecně
- Two of these symptoms are required for the diagnosis, since the complete triad is only observed in about one-third of patients [14]
- Onset is usually after puberty [14]
Edem
- Also affected the genitalia, hands, face, and vocal cords [14]
- Lymphangiograms were interpreted as showing primary hypoplasia of lymphatics [14]
Mutace FOXC2
- Perhaps the most well known of genes responsible for hereditary lymphedema is the FOXC2 gene.
- Identified by University of Michigan scientists and collaborators from the University of Arizona, FOXC2 is the genetic cause of Lymphedema Praecox Meige Syndrome (Hereditary Lymphedema II) and as a causative factor in lymphedema-distichiasis syndrome, lymphedema ptosis and lymphedema yellow nail syndrome. [13]
Literatura:
[8] www.nature.com/ejhg/journal/v16/n3/full/5201982a.html
[13] www.lymphedemapeople.com/wiki/doku.php?id=lymphedema_gene_foxc2
[14] www.ncbi.nlm.nih.gov/omim/153300
Lymfedém a mutace HGF
Lymfedém a mutace MET
Lymfedém a mutace GJC2
Role GJC2
- Gap junctions are implicated in maintaining lymphatic flow [7]
- Missense mutations in GJC2 alter gap junction function and disrupt lymphatic flow [7]
- Until now, GJC2 mutations were only thought to cause dysmyelination, with primary expression of Cx47 limited to the central nervous system [7]
- GJC2 mutations as a cause of primary lymphedema raises the possibility of novel gap-junction-modifying agents as potential therapy for some forms of lymphedema [7]
Mutace GJC2
- Identified six probands with unique missense mutations in GJC2 (encoding connexin [Cx] [7]
Noonan syndrome
- Lymphedem as a part of the syndrome [20]
- Short stature [2]
- Congenital heart defect [2]
- Broad or webbed neck [2]
- Unusual chest shape with superior pectus carinatum, inferior pectus excavatum, and apparently low-set nipples [2]
- Developmental delay of variable degree [2]
- Cryptorchidism [2]
- Characteristic facies [2]
- Varied coagulation defects [2]
- Lymphatic dysplasias are observed with onset at birth or in childhood. [2]
- Pulmonary valve stenosis, often with dysplasia, is the most common heart defect and is found in 20%-50% of individuals. [2]
- Hypertrophic cardiomyopathy, found in 20%-30% of individuals, may be present at birth or appear in infancy or childhood. [2]
Noonan syndrom a mutace
- Mutations in PTPN11 are observed in 50% of affected individuals[2]
- SOS1 in approximately 13% [2]
- RAF1 in 3%-17% [2]
- KRAS in fewer than 5% [2]
- Inheritance is autosomal dominant [2]
Neonatal lymphedema
- Similar to that seen in Turner syndrome, associated with a Y;16 translocation in a male infant.
- Breakpoint in chromosome 16 at 16q24.3 [1]
Literatura:
[1] www.lymphedemapeople.com/thesite/lymphedema_distichiasis.htm
[2] www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=milroy
[7] www.cell.com/AJHG/abstract/S0002-9297%2810%2900213-2
[20] www.ncbi.nlm.nih.gov/pmc/articles/PMC1287178/?tool=pubmed
Lymphatic vessel hypoplasia in fetuses with Turner syndrome
- Turner syndrome is associated with subcutaneous accumulation of fluid in the neck region that can be visualized sonographically from 10–14 weeks of gestation as massively increased nuchal translucency thickness.
- Possible mechanisms for this increased translucency include dilatation of the jugular lymphatic sacs because of developmental delay in the connection with the venous system, or a primary abnormal dilatation or proliferation of the lymphatic channels interfering with a normal flow between the lymphatic and venous systems. [17]
- It seems that the mechanism leading to the increase of fluid in the skin in Turner syndrome is completely different from fetuses with trisomies. [17]
- A microscopic study examining the lymphatic vessels in the skin of spontaneously aborted fetuses with cervical cystic hygromas reported that in non-Turner fetuses, there were numerous dilated lymphatic vessels, whereas in Turner syndrome, there were very few such vessels. In the skin of normal fetuses with no cystic hygromas, lymphatic vessels were evenly distributed [17]
- Investigating women with Turner syndrome and ovarian dysgenesis as a result of a 45XO karyotype lymphangiography revealed hypoplastic lymphatic vessels in the lower limbs, pelvis and retroperitoneal space [17]
- Combination of a characteristic phenotype in females who have one normal X chromosome and either absence of the second sex chromosome (X or Y) with or without mosaicism or partial deletion of the X chromosome. [17]
- The Turner syndrome phenotype includes short stature, stature disproportion, primary amenorrhea, neck webbing, congenital lymphedema of the hands and feet, high-arched palate, short metacarpals, scoliosis, Madelung deformity, hearing difficulties, cardiac and renal anomalies, hypothyroidism, and glucose intolerance. [17]
- Lymphedema in this syndrome affects the extremities and often improves over time. Turner syndrome occurs in 1:2500 to 1:3000 live female births and should always be considered in a female with congenital lymphedema. [18]
Literatura:
[17] humrep.oxfordjournals.org/content/14/3/823.full
[18] www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=milroy
Clinical genetic testing available:
- FLT4, FOXC2, and SOX18 at Prevention Genetics in Wisconsin [5]
- FLT4, FOXC2 at SW Thames Molecular Genetics Diagnostic Laboratory in London [5]
- Both the size of the gene
- Whether a familial mutation (change) has already been identified or if the entire gene must be screened
- Range from $200* to $1500* [5] (Updated: 2/1/10)
Literatura:
[5] www.lymphnotes.com/article.php/id/488/
CCBE1
- Extracellular protein CCBE1
- Collagen and calcium-binding EGF domain-1
- Enhances the lymphangiogenic effects of VEGFC in vivo [Bos et al., 2011] [4]
- Essential for fetal liver erythropoiesis [Zou et al., 2013]
- Important for lymphatic development
- In genetic knock-down screening in zebrafish [Hogan et al., 2009]
Human patients, CCBE1 homozygous and compound heterozygous mutations cause
- Hennekam lymphangiectasia-lymphedema syndrome (OMIM 235510)
- Severe peripheral lymphedema associated with
- Intestinal lymphangiectasias
- Characteristic facial features
- Growth and mental retardation
- Hydrops fetalis [Hennekam et al., 1989; Alders et al., 2009, Alders et al., 2013; Connell et al., 2010] [4]
All CCBE1-mutated patients clearly had Hennekam syndrome
- [Alders et al., 2009]
- Future screens of CCBE1 could be restricted to patients with
- Generalized lymphatic dysplasia
- With or without facial anomalies
- With a recessive mode of inheritance [4]
- Binds to the extracellular matrix and potentiates the effects of VEGF-C on VEGFR-3
- CCBE1 is required for lymphangioblast budding and angiogenic sprouting from venous endothelium
- Humans, homozygous or compound heterozygous mutations that abolish CCBE1 function
- Cause highly penetrant, generalized lymphatic anomalies
- Lymphedema [55]
- Visceral lymphangiectasias [55]
- Associated with typical facial features and mental retardation [55]
- Components of the Hennekam lymphangiectasia-lymphedema syndrome (OMIM 235510) [55]
Možnosti ovlivnění lymfedémů
Stimulace CCBE1
- ?
Synergie s CCBE1
- ?
Galactocerebrosides (GCs)
- The products of Cgt collectively referred to as galactocerebrosides (GCs)
- Major component of the myelin sheaths
- Facilitate the transmission of saltatory conduction (Norton and Cammer, 1984)
Cgt-/- mice
- Display defects in nerve conduction
- Die on postnatal days 18–30 from severe tremor and ataxia (Bosio et al., 1996, Coetzee et al., 1996) [12]
- Defects in postnatal lymphopoiesis
- Specific deficits in stromal elements that support the growth and differentiation of lymphoid precursors (Katayama and Frenette, 2003)
- Fail to mobilize BM HSPCs following G-CSF stimulation
- Deficit is not due to the absence of BM sulfatide
- Likely originates from altered neural influence on osteoblasts [12]
- Adrenergic tone, osteoblast function, and bone CXCL12 are dysregulated [12]
- Number of common lymphoid progenitor (CLP) cells (Kondo et al., 1997)
- Significantly reduced in Cgt-/- mice compared to Cgt+/+ littermates
- Block in lymphoid differentiation (Katayama and Frenette, 2003) occurs before the CLP stage
- Mobilization defect in Cgt-/- mice is unrelated to lymphopenia [12]
www.sciencedirect.com/science/article/pii/S0092867405013280
FOXC2
Funkce
- Forkhead box C2
- Regulates the expression of genes involved in
- Cell growth
- Proliferation
- Differentiation
- Longevity [Fang et al., 2000]
Klinika
- Mutated in puberty or late-onset primary lymphedema associated with distichiasis (LDS, OMIM 153400) [4]
- Hereditary lymphedema II (OMIM 153200) [5]
- Often associated with
- Distichiasis (double row of eyelashes)
- High penetrance
- Not all patients with this feature carry a mutation in FOXC2
- Sometimes ptosis (OMIM 153400)
- And/or yellow nails (OMIM 153300) [5]
- Foxc2–/– mice have
- Abnormal lymphatic patterning
- Arrested lymphatic valve development [5]
- Heterozygotes
- increased recruitment of pericytes hampers the function of collecting lymphatics
- Reminiscent of the human phenotype [5]
Genetika
- Majority of the FOXC2 mutations are
- Insertions,
- Deletions
- Nonsense mutations
- Leading to
- MRNA decay
- Truncated loss-of-function proteins [Dagenais et al., 2004; Ghalamkarpour et al., 2009a; van Steensel et al., 2009] [4]
- FOXC2 suppresses PDGFB production [Shimoda et al., 2011]
- Loss of its activity leads to
- accumulation of vascular smooth muscle cells in collecting lymphatics of knock-out mice and also in patients [Petrova et al., 2004; Norrmen et al., 2009] [4]
- Cca 7% lymfedémů [1]
Diagnostické možnosti v praxi
- ?
Potencionální terapie
Snížení produkce PDGFB v lymfat. cévách
- ?
Potlačení množení smooth muscle cells /pericites in collecting lymphatics
- ?
GATA2
- GATA-binding protein 2
- Zinc finger transcription factor
Funkce
- Putative enhancer element upstream of the key lymphatic transcriptional regulator PROX1
- That is bound by GATA2, and the transcription factors FOXC2 and NFATC1
- Cross-regulation
- Transcription factors modify expression levels of several other proteins
- Some involved in lymphangiogenesis
- Transcription factor that controls PROX1 and FOXC2 expression
- PROX1 regulates FLT4, and FOXC2 controls proteins essential for lymphatic valves, such as connexins [5]
- GATA2 has been most extensively studied in hematopoiesis
- Crucial for hematopoietic stem cell development during both embryogenesis and adulthood
- In hematopoietic progenitor cells
- Gata2 plays key roles during lymphovenous and lymphatic vessel valve formation
Klinika
- Heterozygous germline mutations - range of clinical phenotypes
- Essential roles for GATA2 in the lymphatic vasculature and explain why a select catalogue of human GATA2 mutations results in lymphedema.
Emberger syndrome -Emberger GATA2 missense mutants
- Lymphedema and predisposition to myelodysplastic syndrome/acute myeloid leukemia (MDS/AML)
- Emberger-associated GATA2 missense mutations
- Result in complete loss of GATA2 function
- Regulate the transcription of genes that are important for lymphatic vessel valve development
- Profoundly reduced capacity to bind this element - PROX 1
Conditional Gata2 deletion in mice
- GATA2 is required for both development and maintenance of lymphovenous and lymphatic vessel valves
Mutations in GATA2 linked to a
- Predisposition to myelodysplastic syndrome (MDS, OMIM 614286)
- Acute myeloid leukemia (AML, OMIM 601626)
- Identified in patients with primary lymphedema with myelodysplasia - Emberger syndrome (OMIM 614038) [Hahn et al., 2011; Ostergaard et al., 2011b] [4]
- Monocytopenia with mycobacterial infection syndrome (MonoMAC, OMIM 614172)
- Mostly to: dendritic cell, monocyte, B lymphocyte and natural killer lymphocyte deficiency (DCML) [Kazenwadel et al., 2012] [4]
- Japanese patient with a GATA2 mutation
- MonoMAC and Emberger syndromes [Ishida et al., 2012] [4]
- no obvious genotype-to-phenotype correlations [Hyde and Liu, 2011; Holme et al., 2012] [4]
- Suggesting that modifiers play a role
SNPs in GATA2 have been associated with
- Coronary artery disease
- In arterial development
Ablation of gata2 in zebrafish
- Affects morphogenesis of the dorsal aorta
Gata2 del. or x low expression
- Important for vascular integrity and separation of the blood and lymphatic vascular networks [34]
- Complete loss of function of one GATA 2 allele
- Key factor that predisposes to lymphedema onset
- Clinical heterogeneity in symptoms
Most GATA2 missense mutations incl. prevalent T354M
- Do not correlate with lymphedema
- Hypothesis that complete heterozygous loss of GATA2 function underlies lymphedema was complicated by the description of Emberger syndrome patients with missense mutations
- R361L, C272R , R396Q [34]
Gata2 “null” haploinsufficiency
- Propensity to develop lymphedema
- Through regulation of genes, including Prox1 and Foxc2
- That are important for lymphatic vessel development and valve development
- SiRNA-mediated GATA2 knock-down in primary embryonic mouse LECs
- Reduced PROX1 levels and FOXC2 [34]
Adult heterozygous Gata2?EC/+ mice
- Injected with Evans Blue dye
- Collecting lymphatic vessels of substantially larger caliber than controls
- Reduced transport of Evans Blue dye to the thoracic duct
- Blood within the thoracic duct
Stimulace transkripce/exprese GATA2
Zmírnění lymfedému
GATA2 itself
NOTCH1/RBJ-kappa
- Required to initiate Gata2 expression in hematopoietic stem cells in the embryonic aorta-gonad-mesonephros region
Shear stress
Takže manuální lymfodrenáž a lymfotaping, bandáže
BMP signalling
- Required to induce Gata2
- Shown to control lymphatic vessel valve formation
Inhibice GATA2
zhoršení lymfedému ?
Retinoic acid signalling (Vitamín A !!!)
- Impact on the transcriptional activity of GATA2
- Direct interaction between the zinc fingers of GATA2 and the DNA-binding domain of RA receptor alpha (RAR alpha)
- Aberrant development of LVVs in Cyp26b1–/– mice
- RA signalling is elevated
Mutagenní účinek vysokých hladin vit. A ???
Loss of NOTCH1 results in
- Fewer valves
- Disrupted reorientation of valve endothelial cells
- Reduced levels of valve markers, including
- ITGalpha9
- FN-EIIIA
Notch-induced gene Hes1
- Negatively regulates Gata2 in hematopoietic stem cells of the AGM
- Controlling the production of functional HSC
- NOTCH1 function has recently been shown to be important for lymphatic vessel valve development
Tamoxifen
- Pokud sem správně pochopila, bolkuje funkci GATA2 a v pokusech embryí psů výrazně zhoršoval vývoj lymfatických i venolymatických chlopní a jejich podkoží i krční lymfatické dukty byly naplněny krví
- Zvýšeně citlivá embrya k tomuto účinku byla s již existující mutací v GATA2 [34]
Tedy asi to může být jeden z mechanismů, kterým může Tamoxifen přispívat k rozvoji sekundárního lymfedému např. paže po operaci prsů pro Ca, především u geneticky disponovaných žen
GATA1
- Repress GATA2 expression in hematopoietic cells
- GATA1 is both positively and negatively regulated by the Notch signalling pathway [34]
Další možnosti zmrnění lymfedému
Stimulace PROX1, FOXC2, FLT4
- ?
GJA1 (encoding connexin-43)
Funkce
- Highly enriched on the upstream side of lymphatic valves
- Forms hemi- and intercellular [5]
Klinika
- An amino acid substitution
- Lymphedema, as part of the oculodentodigital dysplasia (OMIM 164200)
- Affects the eyes, face, teeth, and digits [5]
- GJA1 mutation reported to cause
- Lymphedema
- Likely specifically alters channel properties leading to valve dysfunction [5]
- Homozygous inactivation of Cx43 in mice
- Is lethal at birth
- Cardiac malformation [5]
GJC2 (encoding connexin-47)
Funbkce
- CX47 is expressed in
- Lymphatic endothelial cells
- On the upstream side of lymphatic valves
Klinika
- Nonsynonymous mutations in GJC2 were discovered in a few families with
- Late-onset (age 30) autosomal dominant lymphedema (OMIM 613480)
- All 4 extremities (‘four-limb lymphedema’)
- Sometimes associated with
- Saphenous vein insufficiency
- Blepharoptosis
- Involvement of the face or genitalia
- Recurrent cellulitis
- Some families showed reduced penetrance [Ferrell et al., 2010; Ostergaard et al., 2011a] [4]
Genetika
- The GJC2 amino acid substitutions
- Alter but do not abolish connexin function [5]
- Likely cause gain-of-function [4,5]
- Cx47 homozygous knockouts have no lymphatic defect [5]
- Substitutions of highly conserved amino acids in connexin 47 (CX47) cause
- Lymphedema in all four extremities
- Loss-of-function mutations in GJC2
- Found in patients with inherited autosomal recessive Pelizaeus-Merzbacher-like disease (PMLD, OMIM 608804)
- Primarily premature stop codons
- Hypomyelinating disorder of the central nervous system [Uhlenberg et al., 2004] [4]
- Hypomyelinating leukodystrophy 2 (OMIM 608804) without lymphedema [5]
HGF/MET mutations
- In primary lymphedema
- Lymphedema/lymphangiectasia
- Breast cancer-associated secondary lymphedema
- Pathway provides a new target for the prevention and/or treatment of lymphedema [1]
- Confers susceptibility to secondary lymphedema and other syndromic lymphatic variation [1]
IKBKG (NEMO)
Funkce
- An NF-kappa B modulator
- IKBKG to activate NF-?B
- NF-kappaB upregulates PROX1
- Cooperates with it to induce VEGFR-3 expression [5]
- Modulate the activity of transcription factors
- Affect nuclear dynamics
Klinika - mutated in patients
- Complex lymphedema syndromes
- Rare X-linked syndrome
- Anhydrotic ectodermal dysplasia with
- Immunodeficiency,
- Osteopetrosis
- Lymphedema (OLEDAID; OMIM 300301) [5]
- Associated with
- Incontinentia pigmenti in the mother (OMIM 308300)
- Five cases of OLEDAID with an IKBKG mutation have been reported
- Loss-of-function mutations
- Cause incontinentia pigmenti
- Embryonic lethal in males and in homozygous Ikbkg–/– murine females
- Die from severe apoptosis
- OLEDAID-causing mutations are hypomorphs
- Diminish but do not abolish the ability of IKBKG to activate NF-?B
KIF11 encodes EG5
Funkce
- EG5 acts as a homotetrameric kinesin motor
- Kinesin family member 11, a DNA-interacting protein
- Members of this protein family are involved in
- Establishing a bipolar spindle during mitosis for chromosome positioning and centrosome separation [5]
- Mitotic spindle assembly and function [Ostergaard et al., 2012] [4]
- inhibition of EG5 activates the PI3K/AKT pathway [5]
Klinika
- KIF11 mutations discovered to cause
- MLCRD (microcephaly, lymphedema, chorioretinal dysplasia)
- CDMMR (chorioretinal dysplasia, microcephaly and mental retardation)
- Now regrouped as MCLMR (microcephaly with or without chorioretinopathy, lymphedema, or mental retardation, OMIM 152950) [Ostergaard et al., 2012] [4]
- MCLMR can be sporadic or inherited as an autosomal dominant trait
- Mutations are predicted to result in loss-of-function of EG5 [5]
- Heterozygous KIF11 mutations
- Lower limb lymphedema of variable expressivity associated with:
- Microcephaly with or without chorioretinopathy
- Mental retardation (MCLMR; OMIM 152950) [5]
- Kif11+/– mice
- Phenotypically normal [5]
- Kif11–/– mice
- Die prior to implantation [5]
Meige disease - Meige lymphedema
- Genetic disorder
- Lymphedema later develops
- Onset around the time of puberty
- Autosomal dominant disease
- Mutations in the ‘forkhead’ family transcription factor (FOXC2) gene, long arm of chromosome 16 (16q24.3)
- Most common form of primary lymphedema
- About 2000 cases have been identified
- Causes lymphedema of the legs
- Other areas of the body may be affected
- Arms
- Face
- Larynx
- en.wikipedia.org/wiki/Meige_disease
Nonne Milroy syndrom
- VEGFR3
- Od nrozeni
- AD
PTPN14
- An intracellular phosphatase
- Interact with the VEGFC-receptor VEGFR3
- By co-immunoprecipitation upon activation by VEGFC
- Intragenic deletion encompassing both sides of exon 7 of PTPN14 that causes a frameshift p.Ser194Argfs*19.
- protein tyrosine phosphatase
- Non-receptor type 14
- Identified in a consanguineous family with autosomal recessive choanal atresia and lymphedema (OMIM 613611) [Au et al., 2010] [4]
- Dynamic subcellular localization in vitro
- Nuclear in proliferating cells
- Concentrated at intercellular junctions in confluent cells [Wadham et al., 2000; Benzinou et al., 2012]
- Overlap in expression of PTPN14 and VEGFC
- VEGFC stimulation enhances recruitment of PTPN14 to a complex including VEGFR3
- Homozygous deletion of exon 7
- Loss-of-function in a family with juvenile-onset lymphedema
- Choanal atresia
- Developmental delay
- Pericardial effusion in some affected individuals [Au et al.,2010] [4]
- PTPN14 mutations probably explain only a very small proportion of primary hereditary lymphedema [4]
Možnosti ovlivnění lymfedemu ?
Synergie s PTPN14
- ?
Stimulace PTPN14
- ?
Inhibice blokátorů PTPN14
- ?
SOX18 (SRY-box 18)
Funkce
- Hepatocyte growth factor
- High affinity hepatocyte growth factor receptor (HGF/MET)
- Transcription factor
- Important role in early blood vessel modeling [Downes et al., 2009]
- Differentiation of lymphatic endothelial progenitor cells from venous precursors [Francois et al., 2008]
- VEGFR-3 expression is controlled by PROX1
- A crucial transcription factor for initiation of lymphangiogenesis
- PROX1 is under the control of transcription factor, SOX18 [5]
Genetika
- New candidate lymphedema genes [1]
- Mutations in primary lymphedema, lymphedema/lymphangiectasia, and breast cancer-associated secondary lymphedema
- Recessive and dominant mutations in SOX18
- 3 published SOX18 mutations are localized in the
- DNA-binding domain (recessive)
- Transactivation domain (truncating/dominant) [Irrthum et al., 2003] [4]
Klinika
- Hypotrichosis-lymphedema-telangiectasia syndrome (HLTS, OMIM 607823) [4]
- Congenital lymphedema
- Reduced body hair
- Absence of eyelashes and eyebrows
- Localized cutaneous telangiectasias [4]
- Sox18-/- mice exhibit
- Only minor coat defect [Pennisi et al., 2000a] [4]
- Likely due to redundancy with two close homologs, SOX7 and SOX17
- Spontaneous ragged mutant of Sox18 has
- Defective lymphatic and cardiovascular tissues
- Hair follicle defects [Pennisi et al., 2000b] [4]
- A dominant nonsense mutation located in the transactivation domain of SOX18
- May compete for DNA binding without transcriptional activation of target genes
- The human mutations may therefore have dominant-negative effects, via competitive transcription factor binding [5]
- Two recessive substitutions in the DNA-binding domain
- Likely have less affinity to their promoter binding motifs [5]
- Ragged mice, which have Sox18 mutations
- Phenotypically similar [5]
- Sox18 is expressed in endothelial cells, hair and feather follicles, and the heart
- But no cardiac phenotype is seen in patients or mice [5]
Ovlivnění lymfedému
- HGF/MET pathway is causal for a broad range of lymphedema phenotypes
- new target for the prevention and/or treatment of lymphedema
Expression of HGF via plasmid transfer
- Improves lymphedema via promotion of lymphangiogenesis [2]
Stimulace syntézy SOX18
- ?
Synergické působení
- ?
Inhibice blokátoru
- ?
Agonisté jeho receptoru v lymf. cévách
- ?
VEGF and Ang2 in SCLS sera
- But neither the purified immunoglobulin fraction nor sera obtained from patients during remission
- To human microvascular endothelial cells caused
- Vascular endothelial cadherin internalization
- Disruption of interendothelial junctions
- Actin stress fiber formation
- Increased permeability
- Without inducing endothelial apoptosis [18]
- Serum factor(s) present during acute attack phase of SCLS seemed to have an antiapoptotic effect on endothelial cells in vitro [18]
- Clarksone: plasma drawn during an episode from an index case
- Induced a shock-like syndrome when injected into rats
- Contained heparin-precipitable protein [18]
- VEGF and Ang2
- Contribute to transient endothelial contraction in systemic capillary leak syndrome (SCLS)
- In 20 patients with classic acute SCLS
- Serum VEGF and Ang2 levels were significantly elevated in the SCLS group during acute episodes
- Compared with asymptomatic SCLS patients and healthy controls [18]
- Soluble Tie2 and VEGF receptors (VEGFR2)
- May be shed from activated or damaged endothelial cells and inhibit the activity of Ang1-2 or VEGF
- By acting as decoys for the circulating pool
- However, soluble Tie2 and VEGFR2 levels were similar in sera of patients with SCLS and healthy controls [18]
- VEGF and Ang2
- Regulate endothelial barrier function through VE-cadherin
Angiopoietin 2 (Ang2)
- Elevated in episodic SCLS sera
- But not in remission sera
- Ang2 disrupts adherens junctions through myosin light chain phosphorylation [18]
- Ang2 promotes vascular permeability by inhibiting its receptor, Tie2
- Tonic activation otherwise stabilizes VE-cadherin at adherens junctions
- Levels of the Tie2 agonist ligand, Ang1, were similar in symptomatic and asymptomatic patients [18]
- Increases in Ang2 during acute episodes, together with VEGF, could worsen or prolong the leak state [18]
- High levels of circulating Ang2 have been reported in conditions associated with
- Sepsis
- Adult respiratory distress syndrome
- Toxicity induced by therapeutic IL-2 administration [18]
Průběh
- Beginning of the leak phase
- Circulating Ang2 was only slightly higher than basal values
- Increasing at a slower rate than VEGF
- Ang2 levels peaked several days into the hospitalization
- After VEGF levels had normalized
- Peak serum from patient disrupted cytoskeletal and adherens junctions [18]
- Remained elevated during the postleak phase
- Relative to pre-episode values [18]
Antibody-based inhibition of Ang2
- Counteracted permeability induced by episodic SCLS sera [18]
- Ang2 was also elevated in patients with SCLS during asymptomatic periods
- Relative to the control group
- May render them more susceptible to vascular leakage [18]
- Anti-Ang2 pretreatment counteracted the hyperpermeability elicited by these episodic sera [18]
Vascular endothelial growth factor (VEGF)
- Elevated in episodic SCLS sera
- But not in remission sera
- 1983 heparin-precipitable protein was reported: VEGF but in that time was named “vascular permeability factor”
- Ability to induce rapid leakage from blood vessels [18]
- VEGF is secreted by a variety of cells, including:
- Fibroblasts,
- Keratinocytes,
- Mast cells [18]
- Binds:
- Receptor tyrosine kinases expressed on the surface of vascular endothelial cells [18]
- Analogous endothelial pathway regulating vascular barrier function
- Angiopoietin–TEK tyrosine kinase-2 (Ang/Tie2) signaling axis [18]
- VEGF and Angs regulate permeability [18]
- Case report found acutely elevated serum VEGF in one patient with SCLS experiencing active symptoms
- Correlated with the clinical course [18]
- VEGF promotes internalization of VE-cadherin by inducing its tyrosine phosphorylation [18]
- VEGF and Ang2 levels
- Significantly higher in the chronic subset of patients with SCLS than in healthy controls [18]
- Short interval of elevated VEGF (and possibly other permeability factors) may exist in SCLS [18]
- Also stimulates cellular proliferation over time
- Confluent monolayers proliferate, cells become more tightly packed, leading to an increase in electrical resistance
- Not related to barrier function per se
- Interpretation of TEER assays in the presence of anti-VEGF is thereby limited at this point [18]
Průběh
- 3 months before the episode during an asymptomatic period
- Serum and plasma VEGF levels were markedly elevated
- At the start of the episode
- Declined rapidly on admission to intensive care [18]
- Beginning of the “postleak” phase with massive fluid remobilization from tissues into the intravascular space and diuresis
- VEGF levels had already returned to baseline [18]
Anti-VEGF Ab (bevacizumab)
- Yielded less interpretable results
- Probably because of endothelial toxicity of VEGF withdrawal [18]
- Showed no protective effect in SCLS serum, but protected capilaries in focus with added VEGF [18]
Tedy v rámci prevence by se dalo bez obav doporučit i denní konzumace liposolubilního kurkuminu !!!
VEGF
VEGFC
- Vascular endothelial growth factor C
- Major ligand of VEGFR3
- A frameshift mutation
- Identified in 1 family
- Resulting in loss-of-function of the mutant allele [Gordon et al., 2013] [4]
- Reported in one family
- Klinicky indistinguishable from those with a FLT4 mutation
- Allele likely undergoes nonsensemediated mRNA decay in human beings
- Negligible expression of the mutant protein
- Loss of VEGF-C function [55]
Terapeutické možnosti
Stimulace tvorby VEGFC
- ?
Stimulace jeho receptoru - agnonisté VEGFR3
- ?
Stimulace signální dráhy VEGFC - VEGFR3 -...
- ?
Inhibice blokátorů signální dráhy VEGFC - VEGFR3 -...
- ?
FLT4 (VEGFR3)
- VEGF-C/VEGFR-3 signaling regulate expression of genes essential for lymphatic (and venous) valve development and maintenance. These include
- Gap junction proteins
- Connexins [5]
Funkce
- Transmembrane receptor VEGFR3, encoded by FLT4
VEGFR-3 expression
- Controlled by PROX1
- A crucial transcription factor for initiation of lymphangiogenesis
- PROX1 is under the control of transcription factor, SOX18 [5]
- SOX18 mutated in the rare hypotrichosis-lymphedema-telangiectasia syndrome (OMIM 607823) [5]
- Variable-onset lymphedema associated with sparse hair and cutaneous telangiectasias [5]
VEGFR3 signaling
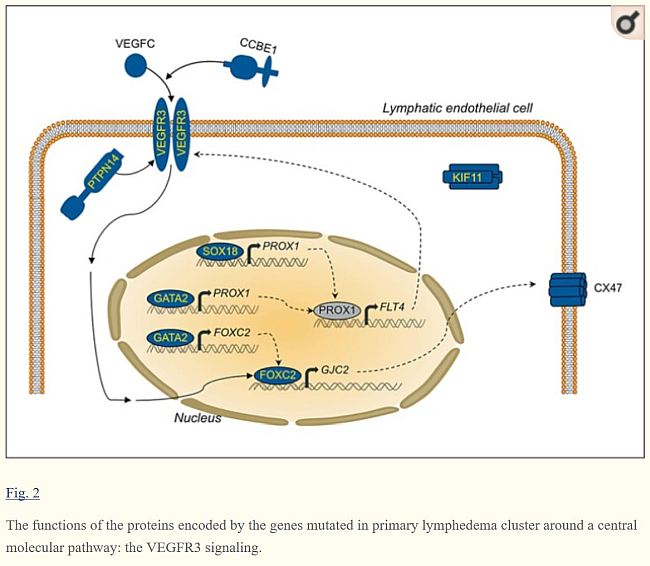
- Theme common to proteins encoded by the 9 genes known to be mutated in patients with primary lymphedema at different levels
- Ligand/receptor (VEGFR3, CCBE1)
- Modification of receptor activity (PTPN14)
- Production of the receptor (SOX18 and GATA2 via PROX1)
- (in)direct control of regulation of transcription (FOXC2, also via GATA2)
- Downstream target gene (GJC2, and FLT4 itself)
- Effects explain more than a quarter of the incidence of primary lymphedema via the
- RAS/MAPK
- PI3K/AKT pathways [5]
Význam pro vznik lymfedemu
- Most of the proteins that are encoded by the genes mutated in primary lymphedema
- Seem to act in a single functional pathway involving VEGFR3 signaling
- Important role this pathway in lymphatic development and function [4]
- Regulation of the VEGFR3 signaling pathway
- Central to the molecular mechanism of primary lymphedema [4]
- VEGFC/VEGFR3 pathway cca 5% lymfedémů [1]
- Dominant VEGFR3 mutations inhibit receptor phosphorylation and prevent downstream signaling. [55]
- The recessive mutation has a weaker effect, resulting in reduced ATP binding [55]
Genetika
- Usually an autosomal dominant disorder
- De novo mutations are not infrequent
- Family history of lymphedema is not required for diagnosis
- Family with a particular recessive VEGFR3 mutation has been reported
- Mutations have been discovered in more than 100 families [5]
Klinické projevy mutace
- FLT4 is mutated in Nonne-Milroy disease
- Congenital bilateral lower limb lymphedema
- All reported mutations localized within the 2 intracellular tyrosine kinase domains of the encoded VEGFR3 receptor [4]
- Cells expressing mutant VEGFR3
- Inhibited autophosphorylation of the receptor
- Indicating downregulation of VEGFR3 signaling [Irrthum et al., 2000; Karkkainen and Petrova, 2000; Ghalamkarpour et al., 2009b] [4]
- Sometimes present with
- Prenatal pleural effusion
- In utero hydrops
- LSG: hypoplasia or aplasia of the lymphatic system is observed [55]
Ovlivnění lymfedemu
- Zvažovali terapii via direct or transgenic delivery of VEGFC [1]
Primární lymfedém
- Rare genetic condition
- Variable in age at onset, expressivity, and penetrance [1]
Autosomal dominant
- Single copy of an abnormal gene is necessary for the appearance of the disease
- Can be inherited from either parent
- Can be the result of a new mutation
- Risk of passing the abnormal gene from affected parent to offspring
- 50 percent for each pregnancy
- Regardless of the sex of the resulting child [3]
Autosomal recessive
Poddruhy of Hereditary Lymphedema
- Congenital hereditary lymphedema
- Hereditary lymphedema, type I
- Lymphedema-distichiasis
- Lymphedema praecox
- Lymphedema tarda
- Milroy disease
- Nonne-Milroy disease
- Noonan syndrome
- Klippel-Trénaunay-Weber syndrome
- Lymphedema-hypoparathyroid syndrome
- Turner syndrome [3]
Mutace
- Mutations in specific genes important to lymphatic development or function
Varying forms of primary lymphedema
- FLT4 (VEGFR3)
- VEGFC/VEGFR3 pathway cca 5% lymfedémů [1]
- Zvažovali terapii via direct or transgenic delivery of VEGFC [1]
- FOXC2 - cca 7% lymfedémů [1]
- SOX18 - +- 0% lymfedémů [1]
- HGF/MET mutations
- In primary lymphedema
- Lymphedema/lymphangiectasia
- Breast cancer-associated secondary lymphedema
- Pathway provides a new target for the prevention and/or treatment of lymphedema [1]
- Confers susceptibility to secondary lymphedema and other syndromic lymphatic variation [1]
Aagenaes syndrome = cholestasis-lymphedema syndrome (CLS)
- Congenital hypoplasia of lymph vessels - causes lymphedema of the legs
- Recurrent cholestasis in infancy
- Slow progress to hepatic cirrhosis
- Giant cell hepatitis with fibrosis of the portal tracts
- Genetic cause is unknown
- Autosomal recessively inherited at chromosome 15q1,2
- Common feature is a generalised lymphoedema from birth or childhood
- Cca one hundred people with this disease are known
- Southern Norway, where more than half the cases are reported
- Parts of Europe and the United States
- Named after Oystein Aagenas, a Norwegian paediatrician.[6]
- Some even experiencing complete liver failure
- Transplantation is necessary
- Cholestasis is accompanied by jaundice, itching, malabsorption of fat and fat-soluble vitamins
- Can lead to skeletal abnormalities and a higher susceptibility to bleeding
- Significant increase of bilirubin concentration in the first months of life, with a later decrease
- Aged 10–20 years
- bilirubin, bile acids and transaminases normal or almost optimal values
- Lower values for total protein, CRP, albumin and creatinine
- Fibrinogen was higher in patients with Aagenaes syndrome
- Improvements
- Nutritional and vitamin supplements, especially in children
- en.wikipedia.org/wiki/Aagenaes_syndrome
Ang2 levels [18]
Distální primární lmyfedem
- Hypofunkce mízních cév
- Kvadrátní prsty
- Firbotizace podkoží
- Papilární linie zvýrazněné
- Max. dorzum nohy
- Od periferie k centru
Distichiasis-lymphedema syndrome
- Rare autosomal dominant
- Multisystem disorder
- Swelling due to fluid accumulation
- Usually around puberty
- Extra eyelashes along the posterior border of the lid margins (distichiasis)
- May range from a few extra lashes to a full set of extra eyelashes
- Swelling most often affects the legs
- Cleft palate
- Droopy eyelids (ptosis)
- Abnormalities of the curved transparent outer layer of fibrous tissue covering the eyeball (cornea)
- Cysts on the spinal cord
- Abnormal sensitivity to light (photophobia)
- Cardiac (heart) defects
- Caused by mutations of the FOXC2 gene [3]
Dsitichiasis syndrom
- často komplikace erysypelem
- Hypopl. lymfedem
- Konjunktivita a fotofobie
- Varixy, vady srdce
Genetika
- Jen asi 5% pac. ostatní secund.
- Vyšetřování vázne
- Prox, TF, růst. f. aj.
- Realtivně vzácné
- Dělení souč. dle věku - popis pouze klinikcé manifestace (zastaralé dělení přes 100 let)
- Insufficience "tam je" od narození ale vznik lymyfedému může být někdy během života
Milroy syndrom
- Chylosni reflux součástí syndromu
Meigova
Angiodyslplazie
Lymfangiomatosy
- S lymforhoeou...
- MTOR inhibitory lokálně off-label s dobrým efektem
- Prušvih s utlakem mediastina
Klipeel Trenaury syndrom
- Otoky v břiše, bolesti, x přijmu potravy, ....
Parkes Weberův syndrom
Proteův syndrom
Nonanové syndrom
Turnerův syndrom
Lymfedema-distichiasis syndrom
Yellov nail syndrom
Melkerson Rosenthalův syndrom
- Crohnova ch, celi....
- Otok čela, fisurovaný jazyk, obrna obličejových svalů
- Dk v normě, ale oblast hlavy problem
Hennekam lymphangiectasia syndrome
- Rare disorder
- Intestinal and renal lymphangiectasia
- Dysmorphic facial appearance
- Mental retardation
- Hypertelorism with a wide, flat nasal bridge
- Epicanthic folds
- Small mouth and small ears
- www.ncbi.nlm.nih.gov/pmc/articles/PMC3193672/
Hereditary lymphedema type IA - Milroy’s disease
Klinika
- Swelling (edema) present at or shortly after birth (congenital)
- Rare cases, edema may develop later in life
- The legs are most often affected
- The extent and location of edema varies greatly
- Genitals may also be affected
- Additional complications sometimes
- Upslanting toenails
- Small warty growths on the affected areas (papillomatosis)
- Abnormally large or prominent leg veins
- In males
- Urethral abnormalities
- Fluid-filled sac along the spermatic cord of the scrotum (hydrocele)
Genetika
- Mutation in the FLT4 gene
- Encodes VEGFR-3 gene
- Located on the 5q35.3 [3]
Hereditary lymphedema type II - Meige disease - lymphedema praecox
Klinika
- Around puberty or shortly thereafter in most individuals
- Most common type of primary lymphedema
- Cca 80 percent of cases [3]
- Lymphedema of the legs
- And other areas of the body may be affected:
- The arms
- Face
- Larynx
- Yellow nails in some people [3]
Genetika
- Mutations of the ‘forkhead’ family transcription factor (FOXC2) gene
- Located on the long arm (q) of chromosome 16 (16q24.3) [3]
- Different mutations of the same gene (FOXC2) disorders include:
- Yellow nail syndrome
- Distichiasis-lymphedema syndrome
- Lymphedema-ptosis syndrome [3]
Incidence primárního lymfedému
- 1,15 na 100 000 u do 20 let
- Počet roste
- 60% pac. hledá lékaře samo pro obtíže trvající i roky
Lymfatické cesty
- Nedostatečný vývoj periferních lymfatických cév a uzlin
- Abnormality v anatomii a funkci lymf. cest
- Poškození periferních lymfatických cév a uzlin mechanicky, opračně, radiačně, chemicky
- Poškození zánětem
- Obliterace a zajizvení po trombóze lymf. cest
- Nedostatečná odolnost vůči poškození
- Nedostatečná regenerace lymf. cest
Lymphedema-ptosis syndrome
- Extremely rare genetic disorder
- Swelling because of fluid accumulation
- Most often affects the legs
- Droopy eyelids (ptosis)
- Usually occurs at or shortly after puberty
- Mutations of the FOXC2 gene
- Autosomal dominant [3]
Lymphedema tarda
- Primary lymphedema
- After the age of 35
- Legs are most often affected
- Arms and other areas may be affected as well [3]
Malformace
- Trunkální forma - primární lymfedem
- Extraturnkální forma - lymfangiomy
- Mikorcysticke
- Makroycysticke
- Venozni
- Zb. emryonální tk.
Inhibitor tkáňového aktivátoru plazminogenu 1 (PAI-1) zvýšený
- Zvýšeně se uvolňuje při výskytu trombózy
- Blokuje aktivaci plazminogenu na plazmin štěpící fibrin na menší, snadno vstřebatelné proteiny
- V kapilárách vlivem PAI-1 nedochází k fibrinolýze
- Vytvářejí se fibrinové manžety způsobující hypoxii a poškození tkáně [12]
To samé se možná děje v lymfatických kapilárách, které při lymfedému časem zanikají ???
Proximální typ lymfedemu
- Atypie uzlin
- Od stehna dolů
- Mohou vést až k elefantiáze
- časně zahájit terapii
Turnerův syndorm
- 45x
- Sodukovitý hrudník
- 30% lymfedem DK
- Pterigium coli
- Vv srdce
Yellow Nail Syndrome
- Hypotrichosis
- Bronchiektazie
- Lymfedem
- Hypolazie
- Slow-growing, hard, yellow, and dystrophic nails
- Respiratory tract disease
- Pulmonary disease, specifically pleural effusion, was added to the diagnostic criteria by Emerson in 1966
- Affects adults over age 50
- Case reports of YNS occurring in children and even newborns
Yellow nail syndrome
- Rare disorder
- Yellow, thickened, and curved nails
- With almost complete stoppage of nail growth
- Loss of the strip of hardened skin at the base and sides of a fingernail (cuticles) may also occur
- Separation of the nails from the nail bed (onycholysis)
- May cause the nails to fall out
- Usually associated with :
- Presence of fluid in the lungs (plural effusion)
- Swelling of the arms and legs (lymphedema)
- Other respiratory problems may occur
- Chronic inflammation of the bronchi and bronchioles (bronchiectasis)
- Chronic bronchitis
- Inflammation of the membranes in sinus cavities (sinusitis)
- Lymphedema usually occurs around puberty
- Mutations to the FOXC2 gene autosomal dominant [3]
- Abnormal accumulation of protein-rich fluid in the interstitial space
Genetika primárního lymfedému
- Cause varying forms of primary lymphedema
- FLT4 (VEGFR3)
- FOXC2
- SOX18
- Hepatocyte growth factor
- High affinity hepatocyte growth factor receptor (HGF/MET)
- New candidate lymphedema genes [1]
- Mutations in primary lymphedema, lymphedema/lymphangiectasia, and breast cancer-associated secondary lymphedema
- Suggests that the HGF/MET pathway is causal for a broad range of lymphedema phenotypes
- New target for the prevention and/or treatment of lymphedema
- Expression of HGF via plasmid transfer improves lymphedema via promotion of lymphangiogenesis [2]
- Cx47 mutations were identified in individuals having secondary lymphedema following breast cancer treatment
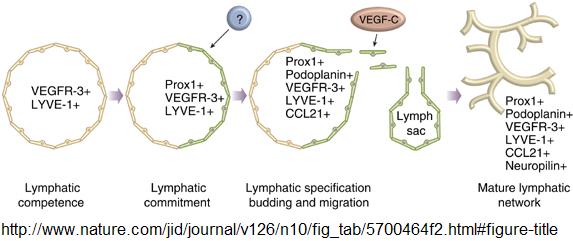


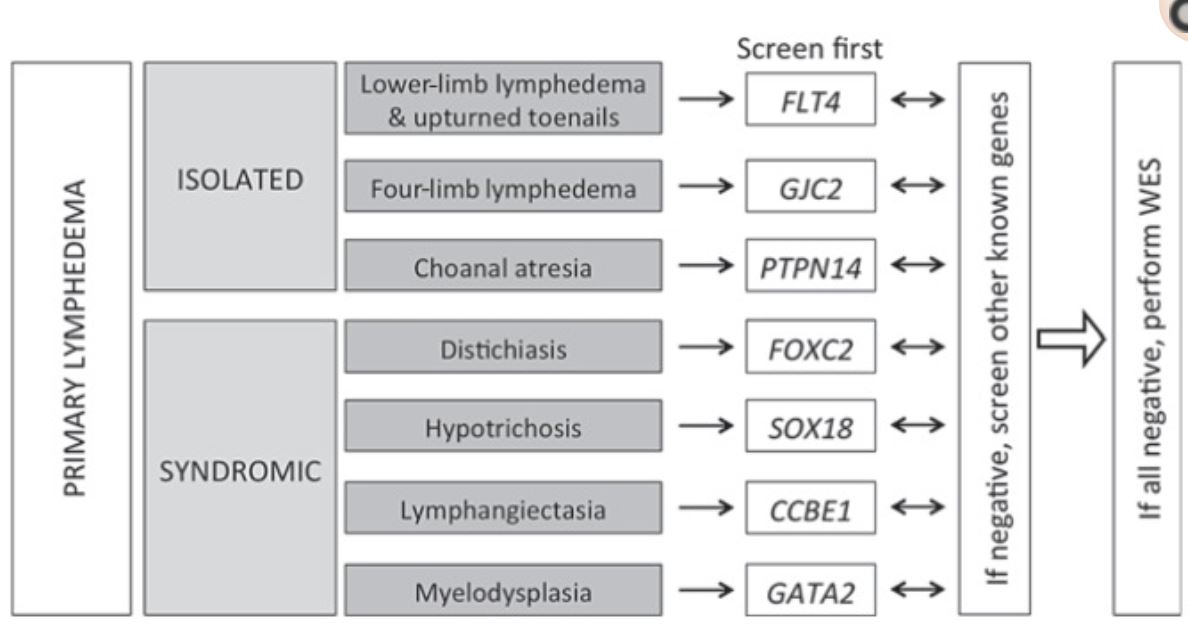
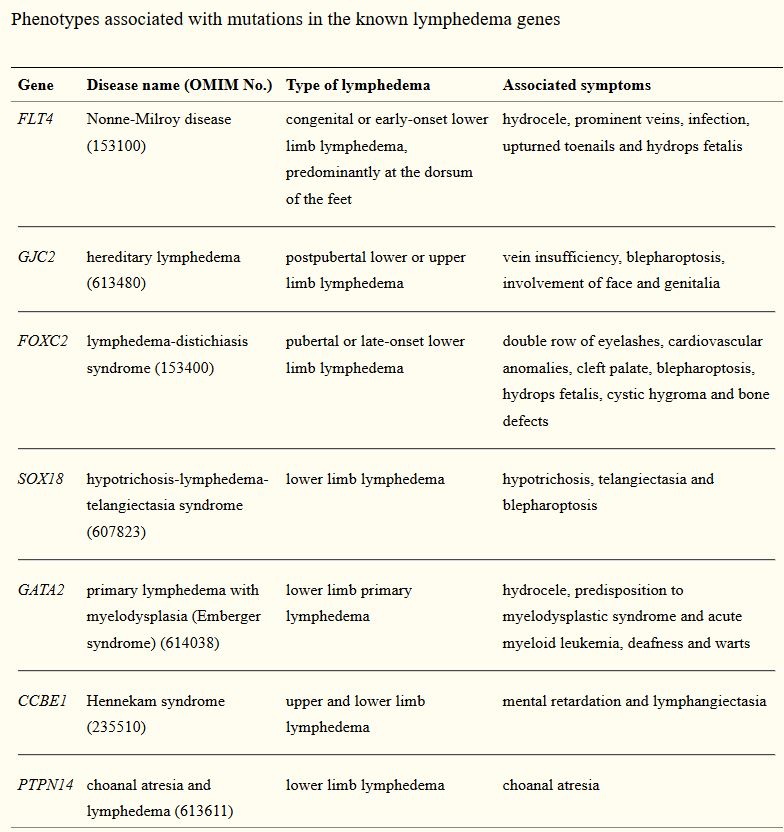
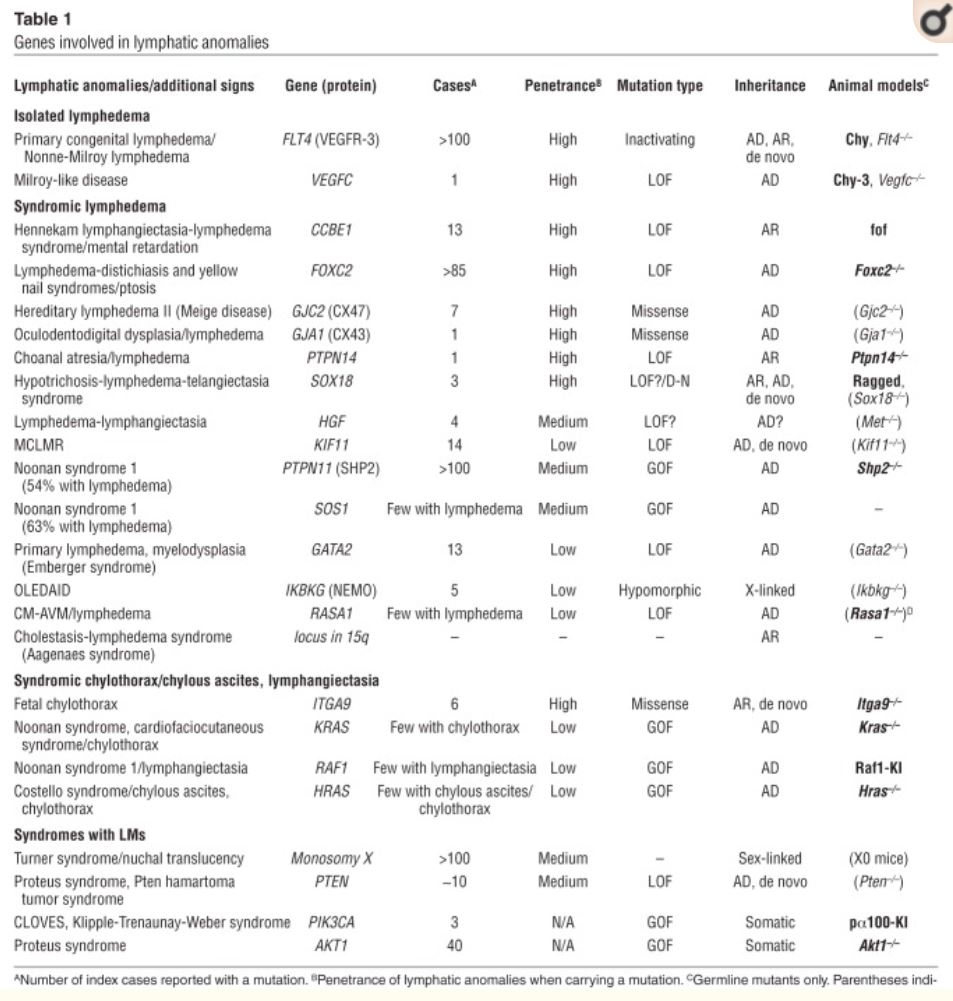
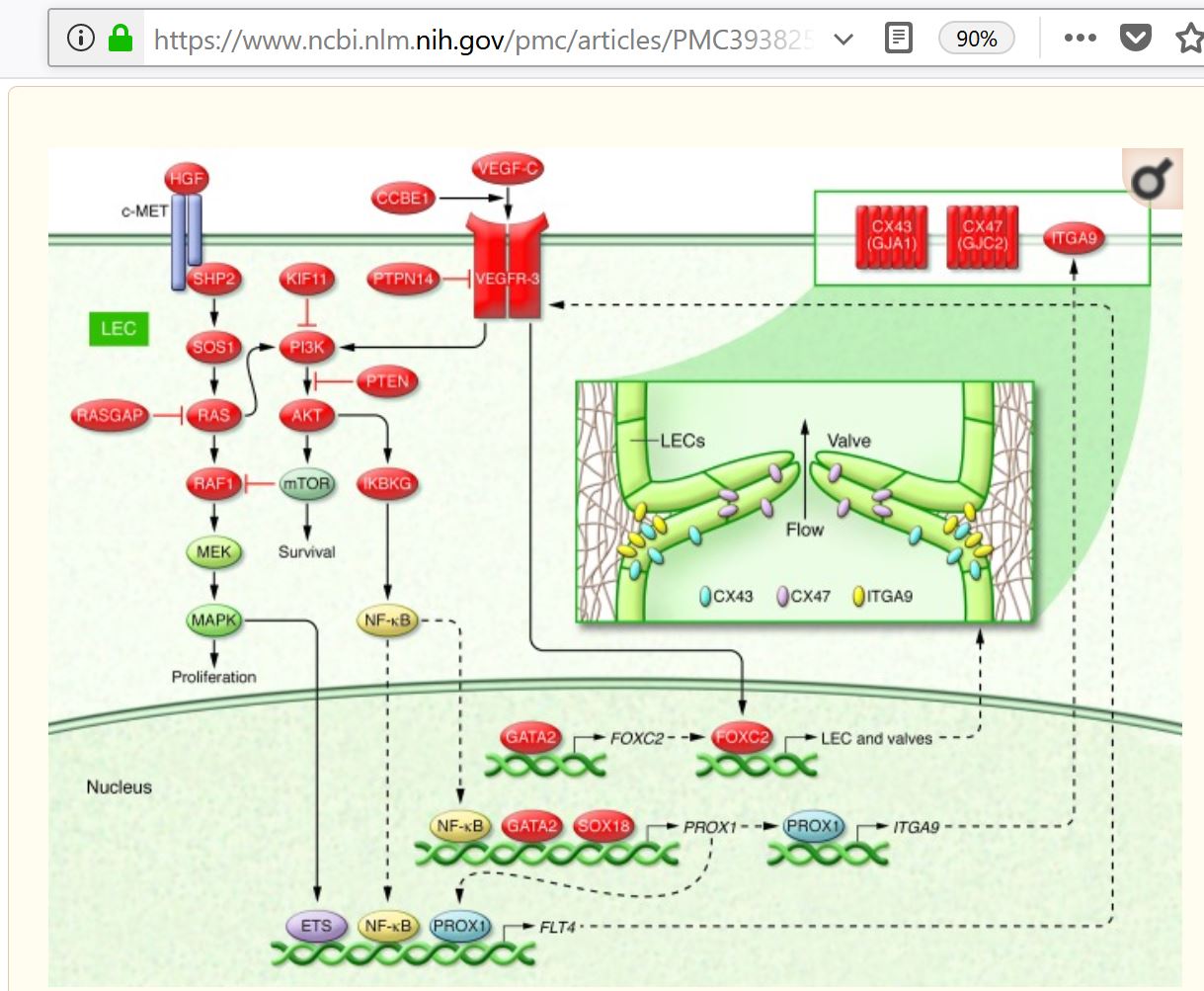

.jpg)
.jpg)
.jpg)
.jpg)