nemoci-sympt/METABOLISMUS/marfanuv-syndrom/dif-dg
- Shprintzen-Goldberg syndrome
- Loeys-Dietz syndrome
- Vascular Ehlers-Danlos syndrome
- Collagen biochemical testing and/or mutation testing:
- TGFBR1
- TGFBR2
- SMAD3
- COL3A1
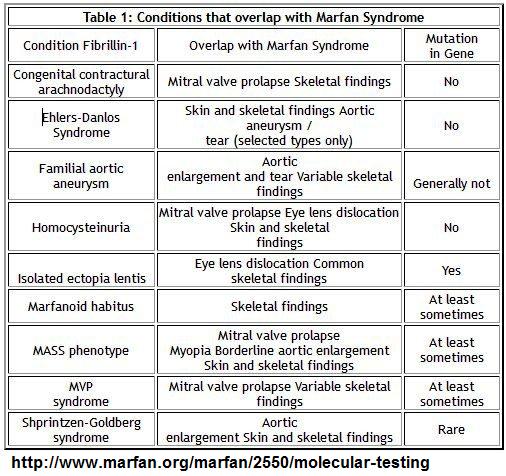
Geneticky příbuzná onemocnění k Marfanovu syndromu
MASS phenotype
- Autosomal dominant
- Heterozygous mutations in FBN1
- Mitral valve prolapse - M
- Myopia - M
- Borderline and non-progressive aortic enlargement - A
- Nonspecific skin and skeletal findings that overlap with Marfan syndrome - S
- In multiple generations in a family
- Intermittent cardiovascular imaging should be maintained
- Difficult to distinguish MASS x Marfan syndrome especially during childhood
- Aortic root Z-score is less than 2.0
- no ectopia lentis
- Systemic score is at least 5
- Only be established for individuals age 20 years or older [1]
Mitral valve prolapse syndrome - MVP
- Autosomal dominant
- Mitral valve prolapse
- Skeletal features reminiscent of the Marfan syndrome
- Can be mutations in FBN1
- Aortic root Z-score is less than 2.0
- no ectopia lentis
- Systemic score is less than 5
- Only individuals age 20 years or older [1]
Ectopia lentis syndrome
- Autosomal dominant
- Ectopia lentis
- Variable skeletal manifestations reminiscent of the Marfan syndrome
- Heterozygous mutations in FBN1
- Unclear whether some individuals show later onset of progressive aortic enlargement
- Intermittent cardiovascular imaging should be maintained
- Aortic root Z-score is less than 2.0
- Patient does not have an FBN1 mutation previously associated with aortic enlargement
- Only individuals age 20 years or older [1]
Autosomal recessive inheritance of isolated ectopia lentis
- Mutations in ADAMTSL4
- Not associated with other manifestations of Marfan syndrome
Shprintzen-Goldberg syndrome (SGS)
- Unclear inheritance
- Dolichostenomelia
- Arachnodactyly
- Pectus deformity
- Scoliosis
- Aortic root enlargement [rare]
- Highly arched palate
- Craniosynostosis
- Developmental delay
- Chiari malformation
- Hypertelorism
- Proptosis
- Rib anomalies
- Equinovarus deformity
- Majority of cases are not caused by mutations in FBN1 [1]
Loeys-Dietz syndrome (LDS)
Loeys-Dietz syndrome types 1 and 2 designate those with and without severe craniofacial involvement [1]- Autosomal dominant
- Long face
- Downward slant of the palpebral fissures
- Highly arched palate
- Malar hypoplasia
- Micrognathia
- Retrognathia
- Pectus deformity
- Scoliosis
- Arachnodactyly
- Joint laxity
- Dural ectasia
- Aortic root aneurysm with dissection
- Dolichostenomelia
- Absent (ectopia lentis)
- Hypertelorism
- Broad or bifid uvula
- Cleft palate
- Learning disability (rare)
- Hydrocephalus (rare)
- Chiari I malformation
- Blue sclerae
- Exotropia
- Craniosynostosis
- Cervical spine instability
- Talipes equinovarus
- Soft and velvety skin
- Translucent skin
- Easy bruising
- Generalized arterial tortuosity and aneurysms
- Dissection throughout the arterial tree
- Aortic aneurysms behave very differently from those in Marfan syndrome
- Frequent dissection
- Frequent rupture at small dimensions
- In early childhood
- Surgical repair has not been complicated by the tissue friability observed in Ehlers-Danlos syndrome, vascular type
- Mutations in
- TGFBR1
- TGFBR2
- SMAD3
- Phenotypic overlap with Loeys-Dietz syndrome [1]
- Strong predisposition for osteoarthritis [1]
Další podobná onemocnění pojivové tkáně
Congenital contractural arachnodactyly (CCA)
- Autosomal dominant
- Marfan-like appearance
- Long, slender fingers and toes
- Heterozygous mutations in FBN2 (encoding fibrillin-2)
- "crumpled" ears, with a folded upper helix
- Contractures of knees and ankles at birth
- Usually improve with time
- Proximal interphalangeal joints also have flexion contractures
- Camptodactyly (palce)
- Hip contractures
- Adducted thumbs
- Club foot may occur
- Kyphosis/scoliosis
- Cca half of all affected
- In infancy and is progressive
- Muscular hypoplasia
- Mild dilatation of the aorta
- Rarely present
- Severe/lethal form
- Multiple cardiovascular and gastrointestinal anomalies [1]
Familial thoracic aortic aneurysms and aortic dissection (TAAD)
- Autosomal dominant
- Cardiovascular disorder
- Similar to that observed in the Marfan syndrome
- Dilatation of the aorta
- Dissections of AO
- At the level of the sinuses of Valsalva
- Ascending thoracic aorta
- Without other phenotypic manifestations
- Mutations in
- MYH11
- ACTA2
- MYLK
- TGFBR2
- May associate with predominant vascular disease
- Some individuals systemic connective tissue disorder
- TAAD and classic Loeys-Dietz syndrome [1]
Ehlers-Danlos syndrome (EDS)
- Group of disorders that have joint hypermobility as a common feature
Classic type
- Autosomal dominant
- Skin hyperextensibility
- Abnormal wound healing
- Smooth, velvety skin
- Cca 50% of individuals mutation in COL5A1 or COL5A2 [1]
Kyphoscoliotic form (previously EDS VI)
- Autosomal recessive
- Kyphoscoliosis
- Joint laxity
- Muscle hypotonia
- Ocular problems
- Risk for rupture of medium-sized arteries
- Respiratory compromise if kyphoscoliosis is severe
- By deficient activity of the enzyme
- Procollagen-lysine
- 2-oxoglutarate 5-dioxygenase 1 (PLOD1: lysyl hydroxylase 1)
- Diagnostic is:
- Increased ratio of deoxypyridinoline to pyridinoline crosslinks in urine measured by HPLC
- Highly sensitive and specific test
- Assay of lysyl hydroxylase enzyme activity in skin fibroblasts
- Molecular genetic
- Gen of enzyme lysyl hydroxylase 1 - on a research basis [1]
Vascular type (previously EDS IV)
- Autosomal dominant
- Joint laxity
- Small joints
- Translucent skin
- Easily visible underlying veins
- Easy bruising
- Wide and dystrophic scars
- Prominent eyes
- Tight or "pinched" appearance
- Organ rupture
- Spleen
- Bowel
- Gravid uterus
- Tendency for aneurysm and/or dissection of any medium to large muscular artery
- no particular tendency for involvement of the aortic root
- This location is not spared from risk
- Tissues can be extremely friable
- Often contributing to surgical catastrophe
- Mutations in COL3A1
- Diagnosis
- Observation of abnormal type III collagen biosynthesis by cultured dermal fibroblasts [1]
Homocystinuria
- Autosomal recessive
- Cystathionine beta-synthase deficiency
- Mutations in CBS
- Variable intellectual disability
- Ectopia lentis and/or severe myopia
- Skeletal abnormalities
- Excessive height
- Limb length
- Tendency for intravascular thrombosis and thromboembolic events
- Life threatening
- Overlap with Marfan syndrome
- Long and lean body habitus
- Pectus deformity
- Scoliosis
- Mitral valve prolapse
- Highly arched palate
- Hernia
- Ectopia lentis
- Cca half of affected responsive to pharmacologic doses of vitamin B6, highlighting the need to consider this diagnosis [1]
Stickler syndrome
- Autosomal dominant
- Connective tissue disorder
- Myopia
- Cataract
- Retinal detachment
- Hearing loss
- Conductive
- Sensorineural
- Midfacial hypoplasia
- Cleft palate
- Alone
- Part of the Robin sequence
- Mild spondyloepiphyseal dysplasia
- Precocious arthritis
- Diagnosis clinically based
- Mutations COL2A1, COL11A1, COL11A2 [1]
Fragile-X syndrome
- X-linked disorder
- Moderate intellectual disability in affected males
- Mild intellectual disability in affected females
- Males may have a characteristic appearance
- Large head
- Long face
- Prominent forehead and chin
- Protruding ears
- Joint laxity - suggest the Marfan syndrome phenotype
- Large testes (postpubertally)
- Autism spectrum disorder, are common
- More than 99% full mutation in FMR1
- Increased number of CGG trinucleotide repeats (>200 typically)
- Accompanied by aberrant methylation of FMR1 [1]