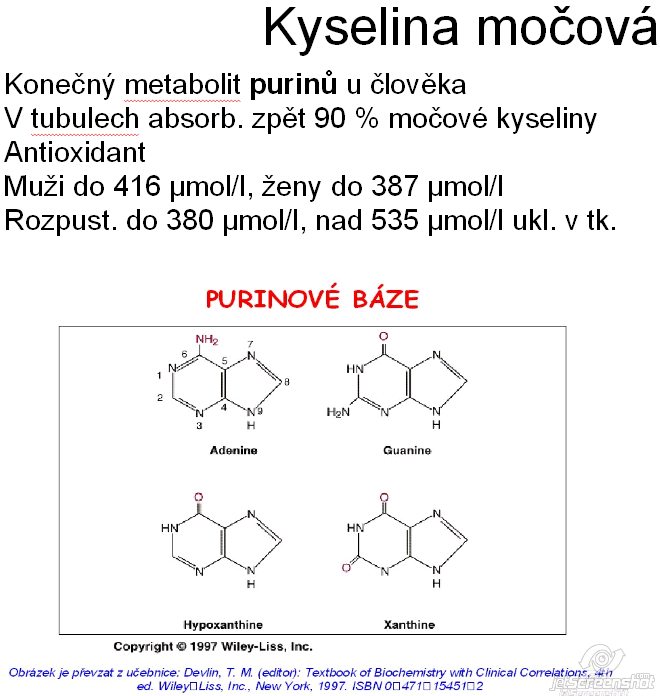
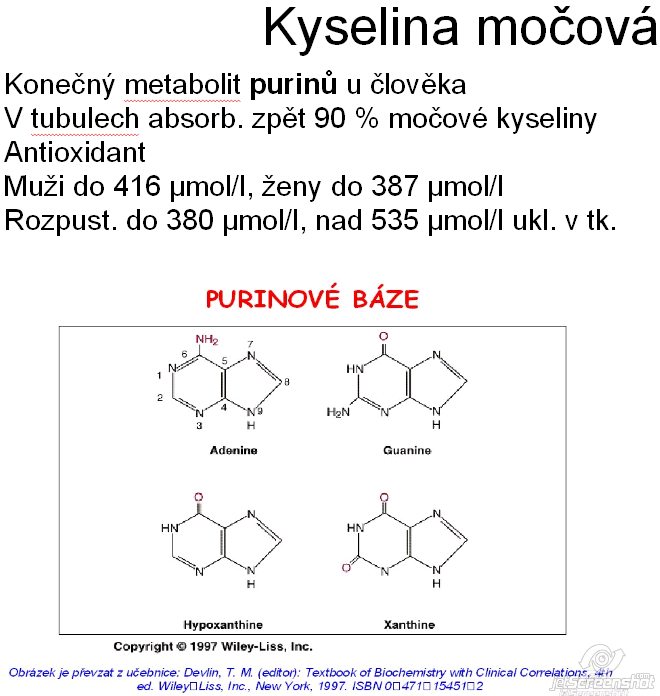
Vlastnosti a role v těle
Vzorec
- 2,6,8 trioxypurine [ C5H4N4O3]
 [1]
[1]- Weak organic acid
- High concentration in extracellular fluid
- Heterocyclic compound
Antioxidační účinky
- Chrání buňky před působením kyslíkovými radikály [4]
- Inhibuje oxidaci askorbové kyseliny [27]
- Ames, 1983
- Jedinci s vyššími hodnotami urikemie mají vyšší inteligenci
- Mají nižší incidenci nádorových onemocnění
- Dožívají se vyššího věku [28]
low UA level has been detected in stroke (8–10), multiple sclerosis (MS) (11,12), infections of the central nervous system (CNS) (13,14) and leprosy reaction episodes (15). As regards IM, the number of previous related studies is limited and the results are conflicting. A total of three early articles (16–18) with small number of recruited subjects and one previous case report (19) were retrieved. Dylewski et al (16) investigated 35 cases with IM after a case report, and reported that 7 men and 2 women had UA levels above the laboratory's upper limit of normal. Cowdrey (17) reported UA elevation during the first 10 days of the disease course in 21 patients. However, Sugita et al (19) described a case of a 27-month-old boy with persistent EBV infection and CNS manifestations, who had lymphadenopathy and low UA levels.
Therefore, the aim of the present study was to analyze the associations between UA and IM in a comprehensive manner, in order to determine whether low UA is a significant risk factor for IM, and whether there is a sex difference.https://www.spandidos-publications.com/10.3892/mco.2017.1433
Modulation of immune responses
Inhibuje oxidací askorbové kyseliny
Konečným produktem
- Metabolismu DNA a RNA
- Volných nukleotidů
- ATP,
- GTP,
- CAMP,
- NAD+,
- NADP
- FAD [27]
Kyselina močová
Kyselina močová je konečným produktem metabolismu purinů u lidí, ptáků a některých dalších organismů. Většina zvířat produkuje alternativní metabolity, jako je močovina. Zvýšené hladiny kyseliny močové v krvi (hyperurikémie) mohou vést k usazování krystalů v kloubech a měkkých tkáních, což způsobuje zánět a bolesti známé jako dna.
Metabolismus kyseliny močové
Kyselina močová vzniká v játrech z purinových bází (adenin a guanin), jež jsou součástí nukleotidů.Enzym xanthinoxidáza je zodpovědný za přeměnu hypoxantinu na xanthin a dále na kyselinu močovou.Normálně se kyselina močová vylučuje ledvinami a malá část i střevní sliznicí.Při narušení tohoto procesu dochází k jejímu hromadění, což vede ke zvýšené koncentraci v krvi a tvorbě krystalů.
Fyziologické a biochemické aspekty
Hladina kyseliny močové v krvi: Fyziologické rozmezí je u mužů 200–420 umol/l a u žen 140–340 umol/l.Při překročení tohoto rozmezí nastává riziko vzniku krystalů urátu sodného, což vede k zánětu a bolesti kloubů.Kyselina močová funguje jako antioxidant, ale ve vysokých koncentracích působí prooxidativně.
Patofyziologie zvýšené hladiny kyseliny močové (Hyperurikémie)
Hyperurikémie může vést k dně, ledvinovým kamenům a dalším metabolickým poruchám.Primární hyperurikémie je geneticky podmíněná, zatímco sekundární může být způsobena nadměrným příjmem purinů v potravě, zhoršenou funkcí ledvin nebo vlivem některých léků (např. diuretik).Krystaly urátu sodného se mohou ukládat v kloubech a vyvolat bolestivý zánět (dnou artritidu), často postihující kloub palce na noze.
Mechanismus vzniku dny
Krystalizace: Kyselina močová ve formě urátu sodného může v podmínkách vyšší koncentrace v krvi vytvářet krystaly.Imunitní reakce: Krystaly jsou v kloubech pohlcovány neutrofily, což spouští zánětlivou reakci.Cytokiny, jako IL-1, TNF-a a další, hrají klíčovou roli v zánětlivém procesu a přispívají k bolesti a otoku kloubů.
Faktory ovlivňující hladinu kyseliny močové
Strava bohatá na puriny (červené maso, mořské plody, alkohol – zejména pivo) přispívá ke zvýšené produkci kyseliny močové.Obezita, hypertenze, inzulinová rezistence a chronické onemocnění ledvin zvyšují riziko hyperurikémie.Léky jako aspirin, cyklosporin a některé diuretika mohou zhoršit vylučování kyseliny močové ledvinami.
Diagnostika hyperurikémie a dny
Stanovení hladiny kyseliny močové v krvi a moči.Klinický obraz (bolest, zánět kloubu, typicky postižení metatarzofalangeálního kloubu palce na noze).Analýza synoviální tekutiny na přítomnost krystalů urátu.
Terapie a léčebné postupy
Farmakologická léčba:
Alopurinol a febuxostat inhibují enzym xanthinoxidázu a snižují produkci kyseliny močové.Probenecid a lesinurad zvyšují vylučování kyseliny močové ledvinami (urikosurika).Kolchicin a nesteroidní antirevmatika (NSAID) jsou používány k úlevě při akutních záchvatech dny.Dieta a změna životního stylu:
Snížení příjmu purinů a alkoholu může pomoci snižovat hladinu kyseliny močové.Hydratace je důležitá pro podporu vylučování kyseliny močové ledvinami.Zvýšení fyzické aktivity a redukce hmotnosti mohou mít pozitivní vliv na hladinu kyseliny močové a prevenci dny.
Syntéza informací umělou inteligencí Chat GPT
Protektivní ve vztahu k Parkinsonově chorobě
- People with gout, and by inference hyperuricemia
- Significantly less likely to develop Parkinson's disease [7]
Konečným produktem metabolismu
- U člověka, opic, plazů a ptáků
- DNA a RNA
- Volných nukleotidů
- ATP
- GTP
- CAMP
- NAD+
- NADP
- FAD [6]
- Formed by the body cca 400 mg/day
- Obtained from food cca 300 mg/day
- Syntéza probíhá v játrech a tenkém střevě
- Obsahují xanthin-oxidázu [30]
- Eliminace probíhá
- Z 80% renálně
- Z 20% intestinálně [30]
- V plasmě se z větší části vyskytuje ve formě solí sodíku a draslíku
- Jen malou část tvoří volná frakce [30]
- Precipitace solí je potencovaná
- Zvýšením poolu kyseliny močové v organismu (hyperurikémie)
- Acidémií (predilekčně periferní tkáně s nižší perfuzí, acidifikace moči) [30]
- Overproduction of ROS
- As a result of xanthine oxidase (XOD) over activity [23]
- Kyselina močová je špatně rozpustná ve vodě
- V plazmě i moči
- Zvyšování její koncentrace v plazmě vede k její krystalizaci
- Následnému usazování ve měkkých tkáních
- Ve formě urátu sodného (Artritis uratica) [2]
- Ve vodě velmi málo rozpustná
- Při pH 7,4 je většina ve formě mononatriumurátu
- Tvoří nasycený roztok (při 37 °C) v plasmě už při koncentraci 420 µmol/l [28]
- 2 dissociable protons with
- PKa 1 cca 5.4
- PKa 2 cca 10.3
- High solubility 120 mg/dL
- Much lower solubility of cca 6.8 mg/dL
- Total uric acid value (dissociated and undissociated forms)
- Solubility limit (6.8 mg/dL)
- Functional pKa1 in serum of 5.75 [13]
- Při pH pod 5,5
- Bývá v moči
- Většina molekul kyseliny močové v nedisociované a tedy méně rozpustné formě
- Může potom vytvářet krystalky, popř. konkrementy
- Rozpustnost urátů velmi nízká - 150 mg/l [5]
- In lower urinary pH
- Increasing in greater proportion its undissociated form
- Because of its poor solubility in aqueous solution [26]
- Ke snížení rozpustnosti přispívá chlad
- Se stoupajícím pH
- se její rozpustnost zvyšuje
- Alkalizací moče na 7,6
- Výrazně stoupá: 1500-2000 mg/l [5]
- Fyziologické pH krve 7,4
- Především v ionizované formě
- S Na+ a K+ vytváří urát sodný či draselný
- Ve vodném roztoku rozpustnější
- Většina ve formě mononatriumurátu [4]
- Tvoří nasycený roztok (při 37 °C) v plasmě už při koncentraci 420 µmol/l [5]
- Most body fluids cca ratio
- Monosodium urate ion - MSU 50 : 1 un-ionized uric acid
- Dominate in extracellular plasma and synovial fluid at pH 7.4 if saturated [23]
- At physiological human pH
- Breakdown of uric acid occurs in 99%
- Forming acid urate ion in the extracellular fluid
- Constant neutral blood pH
- Index between urate ion and uric acid remains stable
- Glucose transporter family isoform Glut9
- Major determinant of plasma uric acid levels and of gout development are identified.
- Metabolic end product of purine metabolism in humans
- Antioxidant properties
- May be protective
- Can also be pro-oxidant, depending on its chemical microenvironment
- Hyperuricemia predisposes to disease through the formation of urate crystals that cause gout
- Hyperuricemia, independent of crystal formation
- Linked with hypertension, atherosclerosis, insulin resistance, and diabetes
- Uric acid, a weak organic acid
- PKa of 5.75
- Principally as monosodium urate (MSU) at physiological pH values
- In humans and the great apes, uric acid is the end product of purine degradation
- In other mammals, it is further degraded into allantoin by uricase
- Mostly found in the liver
- Uricase inactivation
- Urate levels that are much higher in humans (?240–360 uM) in comparison to other mammals (?30–50 uM in mice)
- Common finding in metabolic syndrome
- Inverse correlation between insulin resistance and decreased renal uric acid clearance
- Associated with elevated uricemia
- Obesity, in particular visceral
- Positively associated with hyperuricemia
- Can be reduced by body weight loss
- Frequently observed in patients with cardiovascular diseases
- Association with hypertension and cardiovascular mortality
- May have a direct vascular effect
- Hypertensive children with normal renal function
- Strong correlation between hyperuricemia and blood pressure
- Treatment of these subjects with the xanthine oxidase inhibitor allopurinol
- Significantly lowered blood pressure in a short-term study
- Last 20 years, over 10 studies
- Hyperuricemia independent risk factor for hypertension
- Framingham cohort
- Hyperuricemia was a covariable of other known cardiovascular risk factors for cardiac deaths and coronary heart disease
- Hyperuricemia
- Predisposes to plaque formation and endothelial dysfunction
- Assessed by ultrasonography
- Independent risk factor for cardiovascular mortality
- In patients with established cardiovascular disease
- Elevated urate levels were an independent predictor of cardiovascular events
- Meta-analysis of the association between hyperuricemia and stroke
- Increased risk was found even after adjustment for known cardiovascular risk factors
- Uric acid–mediated vasoconstriction
- Endothelial dysfunction,
- Activation of the renin-angiotensin system
- Hypertension
- Hyperuricemia
- Surrogate marker for early subclinical renal dysfunction
- Cardiovascular complications are secondary
- Powerful antioxidant
- Scavenges singlet oxygen, oxygen radicals, and peroxynitrite
- Chelates transition metals
- Reduce iron ion–mediated ascorbic acid oxidation
- Urate thus accounts for approximately half of the antioxidant capacity of human plasma
- As powerful as those of ascorbic acid
- Can prevent peroxynitrite-induced
- protein nitrosation
- Lipid and protein peroxidation
- Inactivation of tetrahydrobiopterin - a cofactor necessary for NOS
- Uric acid also protects LDL from Cu2+-mediated oxidation
- Underlie the protective effects of uric acid action in cardiovascular diseases, aging, and cancer
- www.ncbi.nlm.nih.gov/pmc/articles/PMC2877959/
- In vitro and cellular studies have nevertheless demonstrated that depending on its chemical microenvironment, uric acid may also be pro-oxidant. For instance, although uric acid can protect native LDL particles against Cu2+-induced oxidation, it also increases the oxidation of already oxidized LDLs, which contain lipid peroxidation products (32, 33), and this dual role appears to depend on the presence of transition metals. As illustrated in Figure ?Figure2A,2A, when uric acid is oxidized by peroxynitrites, urate radicals are produced that could propagate the pro-oxidant state (34), but in the plasma they are rapidly inactivated by reaction with ascorbic acid (31).
- Via Mitogen-Activated Protein Kinase and Cyclooxygenase-2
- uric acid stimulates vascular smooth muscle cell (VSMC) proliferation in vitro.
- Increase VSMC monocyte chemoattractant protein-1 (MCP-1) expression peaking at 24 hours
- Activated the transcription factors nuclear factor-kB, activator protein-1, MAPK
- Increased cyclooxygenase-2 (COX-2) mRNA expression
- Inhibition of p38 (with SB 203580), ERK 44/42 (with UO126 or PD 98059), or COX-2 (with NS398) each
- Significantly suppressed uric acid–induced MCP-1 expression at 24 hours
- N-acetyl-cysteine and diphenyleneionium (antioxidants)
- Inhibit uric acid–induced MCP-1 production
John Kanellis, Susumu Watanabe, Jin H. Li, Duk Hee Kang, Ping Li, Takahiko Nakagawa, Ann Wamsley, David Sheikh-Hamad, Hui Y. Lan, Lili Feng, Richard J. Johnson, (Hypertension. 2003;41:1287-1293.)
Prooxidační stavy při HUA
Rozpustnost a pH
Diprotic acid characteristics
Urate ion
MSU
Ph a rozpustnost
SLC2A9
Uric acid
Hyperuricemia
Another pro-oxidant action of urate has been described during adipogenic differentiation of 3T3-L1 cells (Figure ?(Figure2).2). When these cells are induced to differentiate into adipocytes, addition of uric acid at physiological concentrations further increases ROS production by a mechanism that involves activation of NADPH oxidase (42). This effect in adipocytes may participate in the induction of inflammation and insulin resistance of adipose tissue observed in obesity (43). In vascular smooth muscle cells, uric acid has been reported to stimulate MCP-1 production following activation of NF-?B, MAPKs, and cyclooxygenase 2 (44).
Together, the available information indicates that uric acid has complex chemical and biological effects and that its pro-oxidant or NO-reducing properties may explain the association among hyperuricemia, hypertension, the metabolic syndrome, and cardiovascular disease (45). In addition, when hyperuricemia leads to the formation of microcrystals, it leads to joint and renal inflammation. Chronic inflammation (as in tophaceous gout) leads to bone and cartilage destruction, and chronic hyperuricemia and hyperuricosuria in gouty patients are also frequently associated with tubulointerstitial fibrosis and glomerulosclerosis, signs of local renal inflammation (46). Part of this is explained by the activation of the NALP3 inflammasome to process and secrete IL-1ß (47), but other pathways of inflammation have also been demonstrated (47–49).
There is thus no simple explanation for the possible protective or pathogenic effect of hyperuricemia, and there is clearly a need for more animal models to study this link.
Go to:Urate transporting proteins and genetics of urate transporter pathologiesUrate homeostasis depends on the balance between production and complex processes of secretion and reabsorption in the kidney tubule and excretion in the intestine. It is estimated that approximately 30% of uric acid excretion is by the intestine by mechanisms that have so far not been investigated in detail. Renal mechanisms of urate excretion account for the other 70% and are key to the understanding of hyperuricemia. In patients presenting with gout and primary hyperuricemia, the majority underexcrete urate when the fractional clearance of urate is measured (50). Urate transport by the kidney has been investigated for many years, in part to search for uricosuric drugs to decrease plasma urate levels. So far, several classes of uricosuric drugs have been identified that decrease plasma urate levels, such as benzbromarone, probenecid, sulfinpyrazone, or losartan, whereas other pharmacological agents such as pyrazinoate, the active metabolite of pyrazinamide, nicotinate, and lactate, are antiuricosuric.
In the human kidney, urate handling involves urate glomerular filtration followed by a complex array of reabsorptive and secretory mechanisms taking place in the proximal tubule. In the mouse, both the proximal and distal convoluted tubule appear to be involved in urate reabsorption and secretion, as determined by the localization of the various urate carriers that are discussed below and depicted in Figure ?Figure3.3. It has to be noted that the relative importance of the reabsorption and secretion mechanisms differ among species. Humans, mice, and rats predominantly reabsorb uric acid, whereas pigs, rabbits, reptiles, and birds have more active secretory mechanisms.
URAT1. The urate/anion exchanger URAT1 (SLC22A12 gene) was first identified in a search for organic anion transporter–like (OAT-like) molecules in both gene databases and expression/functional studies in Xenopus oocytes (51). URAT1 is a 12-transmembrane domain–containing protein found in the apical membrane of proximal tubule epithelial cells and transports urate in exchange for Cl– or organic anions. The antiuricosuric agents lactate, pyrazinoate, and nicotinate can serve as substrate for the antiporter activity of URAT1 to increase urate reabsorption. On the other hand, URAT1 is inhibited by the classical uricosuric agents benzbromarone, probenecid, and losartan. Inactivating mutations in URAT1 have been found in Japanese patients with idiopathic renal hypouricemia (51, 52, 53). These patients have plasma uric acid levels lower than 60 µM (or <1 mg/dl) that are associated with urate fractional excretion rates of nearly 100%. These patients are mostly asymptomatic but may develop exercise-induced acute renal failure (54).The mouse URAT1 is 74% identical to the human homolog; it shows similar transport properties, although its Km for urate is higher than that of human URAT1 (1200 µM vs. 370 µM); and it is also located in the apical membrane of proximal tubule epithelial cells (55). Knockout of the Urat1 gene in the mouse leads to increased urate excretion but no significant hypouricemia, indicating that in the mouse, this transporter plays a less important role than in humans for the control of uricemia (56).
OAT4 and OAT10. OAT4 (encoded by the SLC22A11 gene) is a multispecific anion transporter present in the apical membrane of epithelial cells from the proximal tubule (57, 58). It is involved in luminal urate reabsorption by a mechanism that is transactivated by intracellular dicarboxylates but not by the antiuricosuric agents; it is also affected by the diuretic hydrochlorothiazide (59).OAT10 (SLC22A13) is a urate and high-affinity nicotinate transporter expressed in brush border membrane vesicles from proximal tubules and, interestingly, also in cortical collecting ducts in rats (60).
OAT1 and OAT3. The organic anion and urate transporters OAT1 (SLC22A6) and OAT3 (SLC22A8) can function as urate/dicarboxylate exchangers (61–64) and are found on the basolateral side of the same cells that express Oat4 (58). However, Oat3 is also found in all segments of the rat nephron from the proximal tubule to the collecting duct (65). Gene knockout studies in the mouse indicate that absence of OAT1 or OAT3 slightly decreases uricosuria, suggesting that their principal function is in urate excretion (56).Multidrug resistance proteinsMRP4. The multidrug resistance protein MRP4 (ATP-binding cassette family, ABCC4) is present in the apical membrane of proximal tubule epithelial cells. It appears to control ATP-dependent urate extrusion from the cells into the tubule lumen and thus contribute to urate excretion (66–68).ABCG2. Genome-wide association studies for hyperuricemia and gout identified the ABCG2 locus (69). Functional studies (70) demonstrated that ABCG2, which is expressed in the apical membrane of proximal collecting duct cells (71), functions as a urate efflux transporter. Furthermore, a common SNP that introduces a Q141K mutation was found to reduce the transport rate by half when tested in Xenopus oocytes. In a human cohort, the presence of this allele was associated with significantly increased plasma uric acid levels and the risk for gout. The data indicated that at least 10% of all gout cases in individuals of European descent are attributable to this causal variant (69).Other genes identified by genetic association studiesIn the studies mentioned above (69), the sodium/phosphate cotransporter NPT4 (SLC17A3), present in the apical membrane of proximal tubule epithelial cells (72) was also found associated with uric acid levels and gout. This multispecific organic anion transporter is the human homolog of the pig OTAv1 found to be involved in urate efflux (73). In a meta-analysis of over 28,000 individuals (74), several genetic loci were found to be associated with urate plasma levels. These include the SCL17A1 gene encoding NPT1 (a neighboring gene of SLC17A3), URAT1, OAT4, ABCG2, and SCL2A9 (Glut9, see below). In addition, the monocarboxylate transporter MCT9 (SLC16A9), PDZ domain–containing protein 1 (PDZK1), and the protein CARMIL (LRRC16A) were identified. The apical urate transporters URAT1, NPT1, and OAT4 are known to bind to PDZK1 through their C-terminal PDZ domain (75, 76). CARMIL is a protein highly expressed in the kidney and binds actin-capping proteins, thereby increasing actin filament polymerization (77).
The association of PDZK1, NHERF1 (another PDZ-containing protein), CARMIL, and the urate transporters has been suggested to form an apical transportasome complex implicated in the regulation of urate transport (78). This complex also includes the sodium monocarboxylate cotransporters (SMCT1, SLC5A8 and SMCT2, SLC2A12); SMCT1 can also bind PDZK1 (78). Coexpression studies indeed indicated that PDZK1 and NHERF1 overexpression in the presence of URAT1 increases its transport activity (75). These genetic and biochemical data thus indicate that very complex regulatory processes may control the magnitude and direction of urate fluxes across the proximal tubule epithelium. Much more work is necessary to understand them in detail and to determine whether they are regulated by various hormones or metabolic states.
The same meta-analysis (74) also identified the glucokinase regulatory protein (GCKR) locus with hyperuricemia. The role of GCKR in urate transport is, however, unclear. This protein is known for its role in the control of glucokinase activity and glucose utilization by liver, and other genetic association studies have found the GCKR locus to influence triglyceride levels (79, 80).
Glucose transporter family member SLC2A9GLUT9 (SLC2A9) was initially identified by sequence similarity with members of the glucose transporter (Glut) family (81). GLUT9 has the structure of a type II Glut isoform, with 12 transmembrane domains, a large extracellular loop between the first and second transmembrane domains, and both amino- and carboxyterminal ends on the cytoplasmic side (82). In both humans and mice, GLUT9 exists as two alternatively spliced variants that encode different aminoterminal cytoplasmic tails (83, 84). Human GLUT9a has 540 amino acids and is encoded by 12 exons, whereas GLUT9b is 512 amino acids long and encoded by 13 exons, spread over an approximately 250-kb genomic region. In both humans and mice, GLUT9b expression is restricted to liver and kidney, whereas GLUT9a has a broad tissue distribution including liver, kidney, intestine, leukocytes, and chondrocytes (85), where its expression is upregulated by inflammatory cytokines (86) (Figure ?(Figure4).4). In polarized epithelial cells, human GLUT9a is expressed in the basolateral membrane, whereas GLUT9b is targeted to the apical pole (84), and in human kidney, GLUT9 is present in the proximal tubule (84). In the mouse, Glut9 is present in the distal convoluted tubule (83), both in the basolateral and apical membranes (87) (Figure ?(Figure3),3), but it is not yet known which isoform is present in each pole of the cells, in particular since both mouse isoforms have been reported to be targeted to the basolateral membrane of MDCK cells (83).
Functionally, Glut9 was initially reported to be a glucose (88) and/or fructose (89) transporter that, in contrast to other members of the Glut family, could not be inhibited by cytochalasin B. However, the glucose and fructose transport activity reported in these publications was very low and could not be observed in other studies (90, 91). Furthermore, genetic inactivation of the major liver glucose and fructose transporter (Glut2) completely suppressed glucose uptake by hepatocytes (92), even though Glut9 is still highly expressed on their cell surface, indicating that sugar transport is most probably not a physiologically relevant function of Glut9.
Remarkably, the function of GLUT9 was revealed by human genetic studies. Indeed, genome-wide association studies found that the major locus associated with uric acid plasma levels in human cohorts was the SLC2A9 gene, explaining up to 3.5% of serum uric acid level variations (93). This initial study was rapidly followed by several similar reports that replicated, in other cohorts, the observed association of SLC2A9 with uricemia and demonstrated that human GLUT9 is a urate transporter (69, 94–97). Detailed transport studies of the mouse Glut9a and Glut9b splice variants confirmed that the forms have indistinguishable kinetic properties, with a Km for urate of approximately 0.6 mM, and that transport cannot be competed by excess glucose or fructose. Furthermore, transport is electrogenic and independent of Na+ or Cl– concentrations but dependent on membrane potential (90). Importantly, urate transport can be inhibited by the uricosuric agents benzbromarone (90% inhibition of transport) and losartan (50% inhibition) but only marginally by pyrazinoate. The general glucose transporter inhibitor phloretin inhibited transport by approximately 50%, but cytochalasin B was inactive.
Glut9 in genome-wide association studiesIn addition to the association of SLC2A9 with serum uric acid levels, a significant association with gout was reported (95). This study showed a greater impact of the minor allele on decreasing plasma uric acid with higher BMI (98). The SLC2A9 (and ABCG2) SNPs that were associated with gout were, however, not linked with coronary artery disease in the German MI Family Study (99), and no association of the SLC2A9 SNPs could be found with hypertension (96). All studies found a higher impact of the SLC2A9 SNPs in females than in males, and in one study (95) the level of expression of the mRNAs for both GLUT9 isoforms was evaluated in leukocytes, and a significant relationship was found between increased expression of the GLUT9b, but not GLUT9a, isoform and plasma uric acid levels.
Go to:Monogenic mutations in SLC2A9Monogenic forms of hypouricemia have now been linked with mutations in the SLC2A9 gene. Anzai et al. found a P412R mutation in a hypouricemic patient (91); Dinour et al. reported an L75R mutation and a 36-kb deletion present in two different families of hypouricemic patients (100); and Matsuo et al. found two patients with GLUT9 mutations (R198W and R380C). In the latter study, these two patients were identified from a group of 70 hypouricemic patients, 47 of whom had mutations in URAT1 (101). When tested in Xenopus oocyte expression systems, the GLUT9 mutations severely impaired urate transport activity. The individuals with the L75R mutation had mean serum uric acid concentrations of 0.17 ± 0.2 mg/dl and a fractional excretion of uric acid (FE urate) of approximately 150%, a much more severe phenotype compared with individuals carrying a URAT1 mutation. Moreover, a FE urate greater than 100% is suggestive of active secretion of uric acid in the lumen, by an unclear mechanism. Three of these individuals with SLC2A9 mutations had nephrolithiasis, and three had a history of exercise-induced acute renal failure.
Dalmatian dogs excrete large quantities of urate and often develop renal uric acid crystals and nephropathy. They show a defect in uric acid conversion to allantoin, which is not due to a uricase mutation but rather associated with impaired liver urate uptake and renal reabsorption in the proximal tubule (102). Genetic studies have now identified a single mutation of a highly conserved cysteine in transmembrane 5 of Glut9, C188F, as the mutation causing both liver urate uptake and renal reabsorption defects (103). This phenotype is very similar to that observed in mice with genetic inactivation of the Glut9 gene.
Genetic inactivation of Glut9 in mice (104) induces moderate hyperuricemia and massive renal excretion of urate, with a fractional excretion of approximately 100% in males and approximately 150% in females. This is the result of a combined defect in urate conversion into allantoin in the liver and of renal reabsorption. Because Glut9 is also present in the mouse intestine, its genetic inactivation may also prevent intestinal excretion of urate, although this has not been formally tested. These mice display an early-onset nephropathy, characterized by obstructive lithiasis, tubulointerstitial inflammation, and progressive inflammatory fibrosis of the cortex. They also show a mild renal insufficiency, increased water intake, and approximately 5-fold-increased urine volume, with impaired urine concentration capacity.
Selective inactivation of Glut9 in the liver by tamoxifen injection of adult Alb-CreERT2;Glut9lox/lox mice (Lglut9–/– mice) induces severe hyperuricemia, reaching approximately 200 µM in males as compared with a control value of approximately 40 µM and a daily urate excretion rate that was similar to that of the systemic Glut9–/– mice with a fractional excretion of 25%–35%. This was also associated with a urine concentration defect but with only a small increase in urine volume. Together, these data indicate that Glut9 is required for urate access to hepatic uricase and conversion to allantoin and for urate reabsorption in the kidney.
It is also interesting to note that the Lglut9–/– mice showed no nephropathy or kidney morphological abnormalities, even though the daily urate excretion was the same as in the Glut9–/– mice. This suggests that hyperuricosuria-induced nephropathy requires specific conditions, as found in neonates, which have more acidic urine and which cannot compensate increased urine volume by increased fluid intake. Interestingly, the uricase-knockout mice also show hyperuricemia and massive uricosuria and development of nephropathy with accumulation of urate crystals in the kidney (105), confirming that hyperuricosuria present from the time of birth can induce nephropathy.
Impaired urate homeostasis in the Glut9–/– mice is much more severe than in Urat1–/–, Oat1–/–, or Oat3–/– mice (56), indicating that Glut9 is a major regulator of urate homeostasis. Also, it is important to note that in mice, Glut9 is mostly present in the distal convoluted tubule, whereas Urat1 and the other urate transporting proteins are present in the proximal tubule, as in humans (see Figure ?Figure3).3). This suggests that apical and basolateral expression of Glut9 in the same cells may be sufficient for transepithelial urate reabsorption. This also suggests that in the mouse, urate reabsorption takes place in the distal convoluted in addition to the proximal tubule. This has, however, never been tested directly in the mouse to our knowledge. If Glut9 is absent from the proximal tubule, where the other apical urate transport proteins are found, and since Glut9a is so far the only known basolateral urate protein involved in urate efflux, this suggests that another, as-yet-unidentified basolateral urate transporter may exist in the proximal tubule. Indeed, as noted above, Oat1 and Oat3 double-knockout mice have decreased uricosuria, indicating that these transporters are involved in urate secretion, and, on the other hand, Urat1–/– mice have increased uricosuria, indicating that this transporter, and the proximal tubule, must also be involved in urate reabsorption in the mouse. One cannot, however, exclude low levels of Glut9 expression in the proximal tubule that are not detected by immunofluorescence microscopy, because Glut9a mRNA is observed in this tubule segment (90).
Finally, it is not known whether expression of Glut9 in the distal tubule is unique to the mouse or whether it is also present in this nephron segment in other species. This observations nevertheless provides an indication that urate renal handling by Glut9 can proceed independent of the presence of the other urate-transporting proteins, in particular Urat1.
As discussed above, it is not yet established whether uric acid is causally linked with hypertension, atherosclerosis, or insulin resistance because there is a lack of animal models to study the role of hyperuricemia. The availability of mice with genetic induction of hyperuricemia in the adult, such as the Lglut9–/– mice, may be of great help in investigating these associations.
Go to:Future perspectivesThe increasing understanding of urate transport mechanisms sheds light on the causes of hyperuricemia. On one hand, it provides a basis to dissect out the genetic influences on hyperuricemia and to start understanding the complex regulation of urate bidirectional fluxes in tubular cells. On the other hand, it will help researchers achieve a better understand the pharmacological basis for drug action, in relation to drugs that predispose to hyper- or hypouricemia. Indeed, development of uricosuric drugs that act on specific transporters has already started, and it is hoped that this will eventually lead to a wider selection of effective treatments for hyperuricemia. Finally, although efforts have concentrated on the renal transport mechanisms, it is intriguing that they may have a function in other organs or tissues. For example, what is the function of the Glut9 transporter in the leukocytes and chondrocytes? Are transporter mechanisms important in the response of endothelial cells to urate, and can they explain the vascular effects of urate? The availability of mice that are deficient for different transporters will certainly facilitate investigations in the future.
Uric Acid Stimulates Monocyte Chemoattractant Protein-1 Production in Vascular Smooth Muscle Cells