Patofyziologie
Angiogeneze
- Development of aberrant, diverting blood vessels with tumor angiogenesis
- Inability to reach the tumor cells in vivo by chemoterapeutics
- In vitro chemotherapy resistance tests
- Correlate better with lack of activity of chemotherapy agents in the clinical setting
- In vitro chemotherapy sensitivity tests
- Correlate less with the presence of clinical activity
Dormant cells
- A large portion of a tumor doesn’t proliferate
- Sitting there dormant
- Making a mess, and secreting harmful cytokines that affect patients’ performance status
Heterogenita
- Intratumoral heterogeneity
- Genomic and metabolic alterations
- Can use different sources of energy etc.
Šíření
- Adenokarcinom pankreatu
- Typické perineurální šíření (prorůstání do nervů – obvykle nervi splanchnici)
- Bývá zdrojem silných bolestí
- V pokročilejších stádiích do okolních orgánů
- žlučovody, duodenum, cévy [9]
- častý je i rozsev po peritoneu
- Metastazuje
- Do lokálních lymfatických uzlin
- Nodi lymphatici hepatici – z hlavy pankreatu
- Nodi lymphatici coeliaci et pancreaticolienales – tělo a kauda [9]
- Hematogenně
- Do jater
- Později do plic a kostí [9]
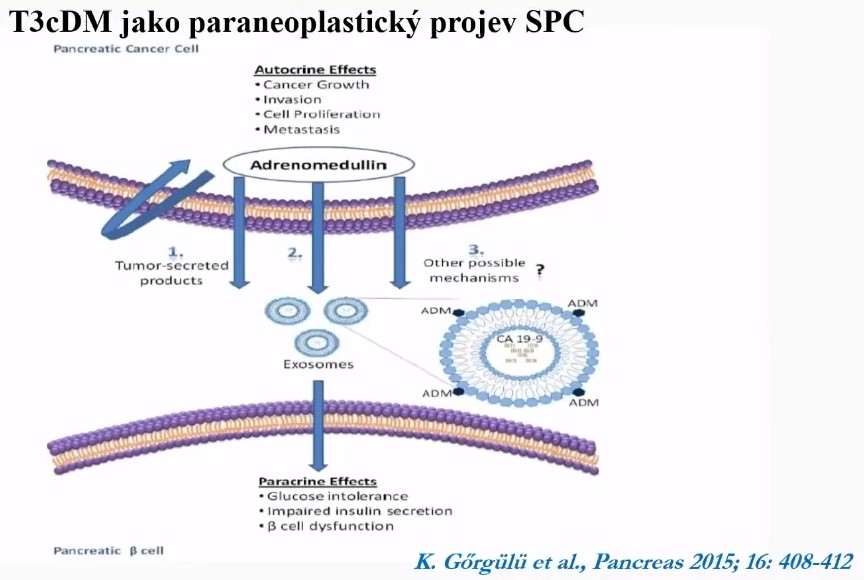
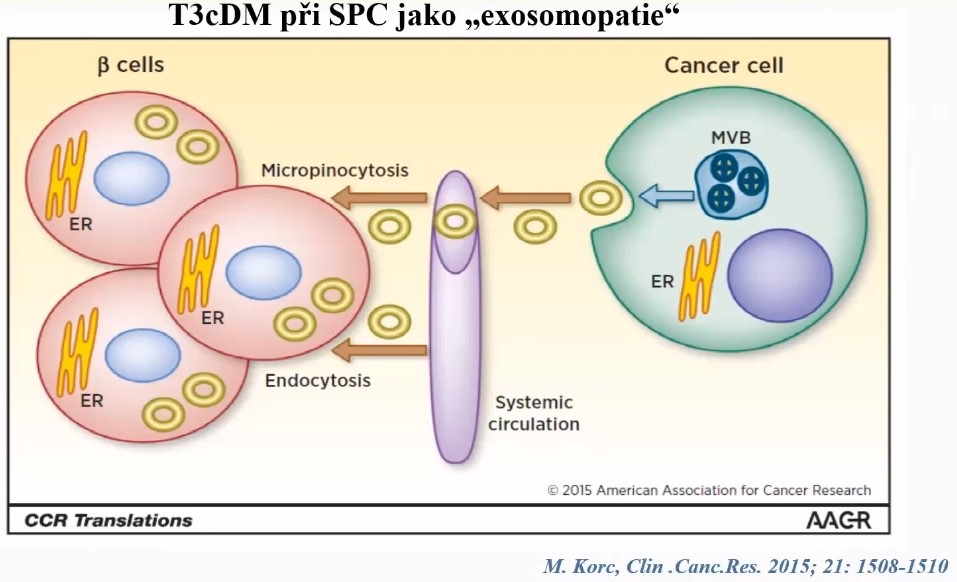
Amylin - islet amyloid polypeptide [IAPP]
Elevated plasma amylin
- In all patients with pancreatic cancer who are diabetic
- Moderately in pancreatic cancer patients with normal glucose tolerance [11]
- Not specific for pancreatic cancer [11]
ATM
Autophagy (macroautophagy)
- Regulated catabolic process of cytoplasmic organelles and macromolecules
- Becomes active under cellular starvation
- Facilitate recycling of cellular material for energy production
- Conflicting implications [18]
- Autophagy induction
- Found to correlate with tumorigenesis suppression in:
- Breast cancer
- Mantle cell lymphoma
- Chronic myeloid leukemia
- Non-small cell lung carcinoma
- Cervical cancer [18]
- Tumor cells autophagy as pro-oncogenic role in transformation
- In pancreatic cancer
- Induced by oncogenes
- In subsequent tumor maintenance
- Basal autophagy
- Elevated in all examined human-derived PDAC cell lines
- In 81% of primary PDAC tumor samples
- In all high-grade pancreatic intraepithelial neoplasms [18]
- Minimal autophagic signaling was observed in:
- Low-grade PanIN
- In normal pancreatic ductal epithelium [18]
- Prominent presence of LC3
- Positive correlation between heightened autophagy and increased tumorigenic progression
- Correlation between autophagy induction and oncogenic constitutively active Ras
- Need for autophagy in Ras-induced malignant cell transformations
- In cancers mediated by Ras oncogenes
- Cancer cells develop autophagy addiction as a survival mechanism [18]
- Oncogenic K-Ras and H-Ras
- Shown to promote autophagy
- Supports transformation
- Cell survival [18]
Induction of autphagy
- Dense stroma
- Poor vascularization
- Hypoxic conditions
- Low nutrient intake
- Weak growth factor flux
- Induce autophagy
- By the adaptive response action of the chief transcriptional regulator HIF-1? [18]
- Activates:
- Transcription of Bnip3 and Bnip3L
- Essential for hypoxia-induced autophagy
- Compete with Beclin 1 for binding to Bcl2
- Releases Beclin 1
- Causing induction of autophagy
- Expression of Bnip3
- Negatively correlated with the progression of pancreatic cancer
- Unlike other HIF-1alfa target genes [18]
- Bnip3 was silenced by gene methylation in PDAC tumor cells
- Becomes downregulated as the disease progresses
- Alternative mechanisms of continuous induction of autophagy at the more advanced stages of disease [18]
Invagination of a single-membrane vesicle
- MTOR activity
- ULK1 and ULK2 proteins
- Activity of the ULK1/2-Atg13-FIP200-Atg101 complex [18]
- Mediates delocalization of class III PI3K (PI3KC3) from microtubules to the endoplasmic reticulum
- Initiation of vesicle nucleation [18]
- PI3KC3 complex contains the autophagy proteins Beclin1, p150 and Ambra 1 [18]
- Generates phosphoinositide 3-phosphate in the nucleation membrane
- Stimulates recruitment of other autophagy (Atg) proteins to the autophagosome [18]
- Atg12 is conjugated to Atg5 and Atg16L1
- Through an ubiquitination-like process
- Form the Atg16 complex
- Mediates expansion and progression of autophagy at the autophagosomal membrane [18]
- Additional ubiquitination-like process
- Most specific step of autophagy
- Atg4 cleaves LC3 protein
- Exposing its C-terminal glycine
- Binds to phosphatidylethanolamine (PE)
- Promoting recruitment of LC3-PE to the autophagosomal membrane [18]
Sequesters cytoplasmic components into a double-membrane vesicle
Forms the autophagosome
Fused to the lysosomes
Lysosomal degradation [18]
Inhibition of autophagy
- ROS induction
- Augmentation of DNA damage
- Impaired mitochondrial oxidative phosphorylation
- Growth suppression of pancreatic cancer cells
- Tumor regression
- Prolonged survival in a K-Ras-driven genetic mouse model of PDAC
- Autophagy is necessary for tumorigenic growth of pancreatic cancer
Autophagy inhibitors
- Chloroquine
- Genetic intervention
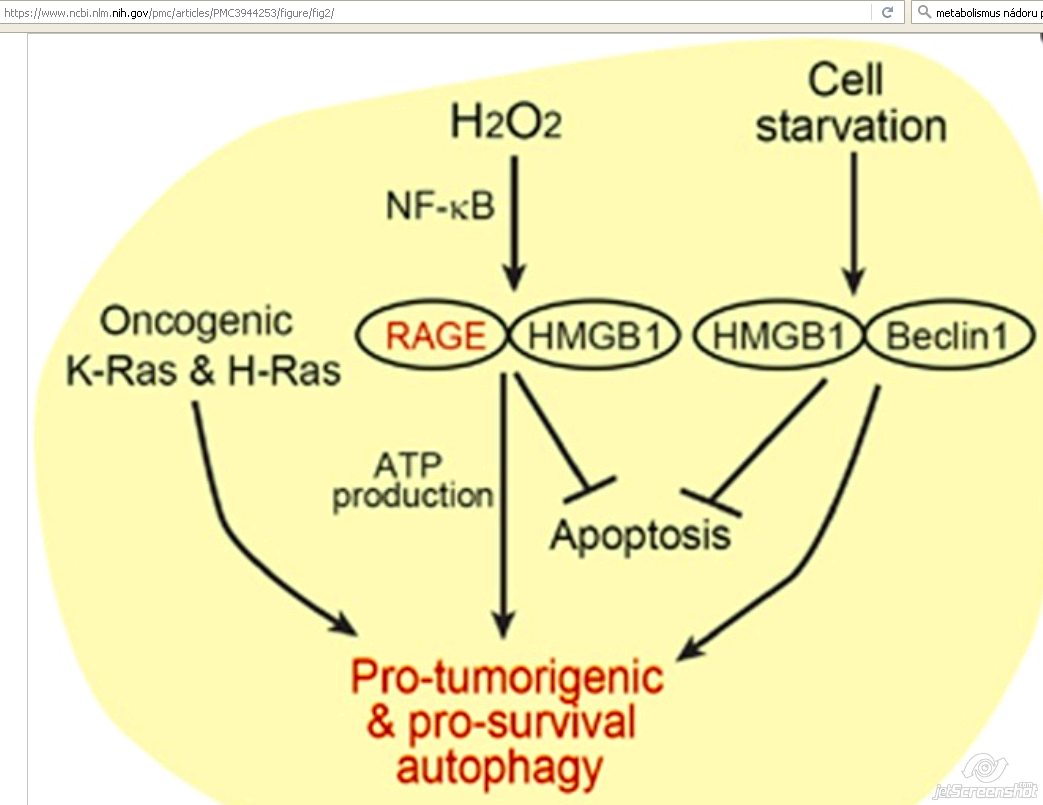
RAGE - receptor for advanced glycation end products and ROS
- Prominent factor in autophagy in PDAC !!! [18]
- Multiligand receptor of the immunoglobulin superfamily
- Intracellular generation of ROS
- Tumor-promoting inflammation
- In PDAC tumor cells, RAGE is overexpressed
- Correlated with:
- Tumor cell survival
- Migration
- Invasiveness [18]
Depletion of RAGE
- Significantly increased sensitivity to:
- Hypoxia
- UV radiation
- Cytotoxic chemotherapy
- Increased induction of cleaved caspase-3
Absence or reduction of either RAGE or HMGB1
- Significantly reduced ATP production
- Slowed tumor growth [18]
RAGE overexpression
- Enhanced cell survival
- Reducing apoptosis
- Promoting autophagy
- Binding several ligands:
- Nuclear chromatin remodeling protein = high-mobility group box 1 (HMGB1)
- Extracted from necrotic and inflammatory cells
- In the extracellular environment contribute to inflammation and tumor progression
- Decrease in intracellular HMGB1 through targeted knockdown
- Reduce autophagy
- Increase the sensitivity of PDAC-derived cells to:
- Apoptosis induced by the chemotherapeutic drug melphalan
- Interaction of HMGB1with RAGE leads to:
- Enhanced cell resistance to programmed cell death in PDAC
- Increase RAGE expression
- Caused by exposure of pancreatic tumor cells to H2O2
- Via activation of the NF-?B signaling pathway
- Inflammatory pathway mediated by HMGB1 and RAGE
- Essential for optimal mitochondrial production of ATP
HMGB1
- Also shown to mediate autophagy in human Panc and mouse Panc02 pancreatic carcinoma cell lines
- Interactions with Beclin 1
- Cellular starvation
- Oxidated HMGB1 translocates from the nucleus to the cytoplasm
- Disrupts interactions between Bcl-2 and Beclin 1
- Competitively binding to the Beclin 1 [18]
BCL2
B-Raf
- Serine/threonine protein kinase
- Located second to Ras in the signaling cascade
- Common mutational pattern in a few primary cancers
- 10% of colorectal carcinomas
- 66% of melanomas
- Mutations in K-Ras and in B-Raf are nearly always mutually exclusive
- Appear in pancreatic cancers with wild-type Ras
- At a rate of one in every three cases [18]
BRCA2
CDKN2A/2B tumor suppressor locus encodes endogenous CDK4/6 inhibitors
CDK4/6 activity
- Controlling gluconeogenesis
- Responsiveness to insulin [17]
Loss of the CDKN2A/2B tumor suppressor locus (CDK4/6 inhibitors)
- One of the hallmark genetic events in PDA
Cyclin-dependent kinase 4/6 (CDK4/6) inhibitors
- Cytostatic
- Preventing cancer cells from growing and dividing [17]
- Induce cyclin D1 protein levels
- RB activation was required
- Sufficient levels of RB for mitochondrial accumulation [17]
- + glycolytic metabolism
- + glycolytic intermediates
- + glucose 6-phosphate
- + fructose 1,6-bisphosphate
- + pyruvate
- + lactate [17]
- Increased lactate efflux
- Measure of the end product of glycolysis [17]
- Increase in media acidification [17]
- + TCA metabolites
- + malate, fumarate, succinate, and alpha-ketoglutarate
- Principally derived from glutamine !! [17]
- Majority of mitochondrial-derived metabolism is fueled by glutamine [17]
- + oxidative phosphorylation [17]
- Via RB pathway [17]
- + accumulation of ATP [17]
- + mitochondrial number
- + reactive oxygen species (ROS) [17]
- + total ROS
- + mitochondria-derived ROS [17]
- + genes involved in peroxisome biosynthesis
- + expression of ROS scavengers:
- + hemeoxygenase 1 (HO-1)
- + catalase (CAT) [17]
- Enhanced glutamate secretion
- Product of glutamine metabolism [17]
- Protected cancer cells selectively against the effect of acute glucose withdrawal [17]
- Enhanced glutamine metabolism was sufficient to rescue the reliance on glucose [17]
- Limited the acute toxicity of mitochondrial inhibitors
- Phenformin
- Rotenone [17]
- Downregulation of phosphorylated RB and E2F [17]
- Compensatory activation of MTOR [17]
- Increased lysosome-associated MTOR [17]
- V.s. TORC1 complex activation [17]
- Increased phosphorylation of ribosomal protein S6 at Ser235/236 [17]
- MTOR complex 1 (TORC1) substrate
- Increased phosphorylation of RSK at Ser 389 [17]
- no increase in either ERK or AKT phosphorylation [17]
- Reduction in Ki67 [17]
- Induction of genes associated with:
- Glycolysis
- Lysosome
- Pyruvate metabolism
- Fatty acid metabolism
- PPAR signaling [17]
- Many of these processes are activated downstream of MTOR [17]
- Cessation of the CDK4/6 inhibition
- Could elicit rapid cell-cycle progression [17]
- CDK4/6 inhibition yields increased metabolic activity
- That is further exaggerated by MTOR activation [17]
CDK4/6 inhibition and MEK inhibition
- Enforced profound cell-cycle inhibition
- Potent cytostatic effect
- Evidence of induced senescence (SA-beta-Gal) [17]
x HO-1 or CAT + CDK4/6 inhibition
- Elicited a significant reduction in PDA cell growth [17]
Cyclin D1
- Requisite activator of CDK4/6 [17]
- Coordinate metabolism and mitochondrial function [17]
Cyclin D1 depletion
- Had little effect on mitochondrial accumulation [17]
Dělení buněk +
- Glucose and glutamine into anabolic pathways [15]
Desmoplastic stroma
- Characteristic of PDAC [15]
Diabetes mellitus or impaired glucose tolerance
- Occurs in up to 80% of patients with pancreatic cancer
- At the time of cancer diagnosis [11]
- Hyperglycemia and stimulation of pancreatic cancer cell growth
- Hypoxic MiaPaCa-2 pancreatic cancer cells with excess glucose results in:
- Increased expression of hypoxia-inducible factor (HIF)-1?
- Increase in cellular ATP
- Decrease in mitochondrial activity [18]
- Glucose metabolism could also be stimulated by:
- Extracellular glucose
- Hypoxia
- Independently of HIF-1? [18]
- Hypoxic pancreatic cells harboring HIF-1? showed:
- An increased capacity for migration [18]
- Glucose
- Stimulates pancreatic cancer cell migration
- HIF-1?-dependent
- HIF-1?-independent [18]
- Na řadě nádorových buněk:
- Receptory pro inzulin
- Insulin-like growth factor (IGF)
- A-izoformy inzulinového receptoru
- Mohou stimulovat mitogenezi i v buňkách, které mají deficit IGF-1 receptorů
- Mohou stimulovat proliferaci nádorových buněk a jejich metastazování [26]
- Postreceptorové děje po stimulaci:
- Inzulinových
- IGF-1 receptorů [26]
- Fosforylaci insulin receptor substrate, IRS
- Mitogen-activated protein kinázu (MAP kinázu)
- K aktivaci signální cesty stimulující buněčnou proliferaci
- Snižující signály pro buněčnou apoptózu
- K růstu i šíření nádoru [26]
- K buněčné proliferaci
- K akceleraci zánětu a aterosklerózy [26]
Hyperinzulinemie :
- +buněčný růst
- + proliferaci
- + diferenciaci
- + kancerogenezi
- + přežívání buněk
- + mitogenezi [26]
- Při inzulinové rezistenci a kompenzatorní hyperinzulinemii
- Inzulinová rezistence oslabuje „kompetitivní“ postreceptorovou cestu
- Fosfatidylinositol-3 kinázu (PI-3 kinázu)
- Aktivaci glukózových přenašečů [26]
- Snížení jaterní produkce vazebného proteinu pro IGF-1
- Zvýšením podílu volné frakce
- Mitotické a antiapoptotické aktivity IGF [26]
- Snížení produkce vazebných proteinů pro pohlavní hormony
- Zvýšení podílu jejich volných frakcí
- Asociovány s vyšším rizikem vzniku postmenopauzálního karcinomu prsu a endometria [26]
- Zánětlivé cytokiny uvolňované z tukové tkáně
- Mohou stimulovat růst i přežívání nádorových buněk
- IL-6
- Vedl k transformaci buněk karcinomu prsu ve více invazivní
- Ovlivnil antitumorózní imunitní reakci [26]
Synthetic glucose analog - 2-deoxy-D-glucose (2-DG)
- Potent metabolic inhibitor
- Selectively directed to tumor cells that consume glucose at high rates under hypoxic conditions
- 2-DG competes with endogenous glucose for key glycolytic enzymes
- Reducing metabolism rate [18]
2-DG + anti-glycolytic agent 3-bromopyruvate (3-BrPA)
- Antitumorigenic effects in MiaPaCa2 and Panc-1 pancreatic cancer cells
- Energy depletion
- Increased cell necrosis [18]
2-DG + metformin
- In LNCaP, P69, PC-3 and DU145 prostate cancer cells
- Leads to almost complete cell-cycle blockage
- Apoptotic cell death
- By inhibition of mitochondrial respiration and glycolysis [18]
Salirasib + 2-DG
- In Panc-1 pancreatic carcinoma cells
- Inhibition of additive cell growth
- Synergistic apoptosis
- Complete contraction of Panc-1 tumor in nude mice [18]
DNA aneuploidy
- Moderate to strong nuclear staining
- 50% of the primary pancreatic tumors [13]
DNA triploidy
- Associated with:
- Mutated Ki-ras gene (p < 0.05) [14]
- Double mutations of c-Ki-ras and p53 (p < 0.05) [14]
FANCG
Faty acids
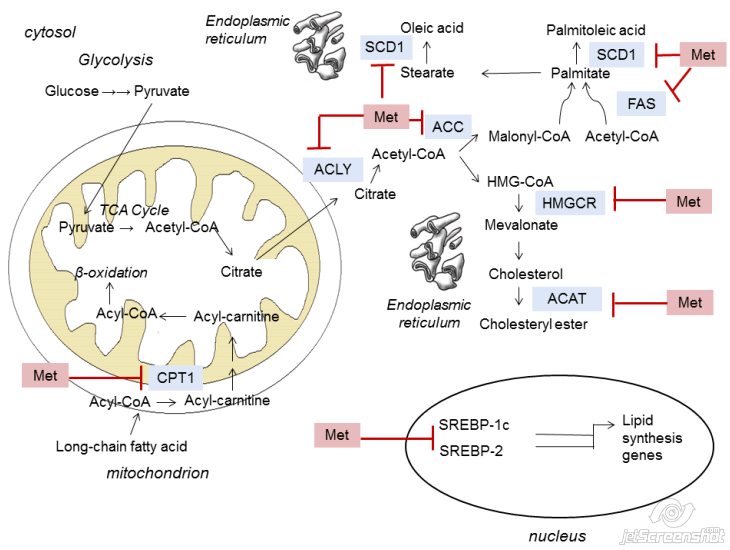
- ATP citrate lyase (ACLY) converts citrate back to acetyl-CoA
- Acetyl-CoA carboxylase (ACC) catalyzes the carboxylation of acetyl-CoA to malonyl-CoA in an ATP-dependent manner
- Acetyl-CoA and malonyl-CoA are then substrates for the production of palmitate
- Seven enzymatic reactions catalyzed by FAS
- In cancer, de novo fatty acid (FA) synthesis is up-regulated
- Mainly for membrane production
- FA for phospholipids
- Post-translational modification of proteins [21]
- ACLY, ACC and FAS expression and activity are upregulated in cancers
- Including pancreatic cancer [21]
- cholesterol and lipid metabolisms
- Linked to cellular transformation [21]
- High HMGCR mRNA levels
- Correlated with poor patient prognosis and reduced survival [21]
- Lvels of mevalonate (MVA) pathway genes
- Significantly correlated with poor prognosis of breast cancer patients [21]
- lipids can stimulate cell growth of human pancreatic cancer cell lines
- Did not enhance the growth of nonpancreatic cell lines
- Normal pancreatic cells utilize lipids as an energy source
- Oxidation of palmitic acid at higher rates than adipose or liver tissue [18]
- FA provide an energy source for pancreatic cell lines
- Proliferation by hormones and growth factors
- Coupled with generation of lipids and lipid-derived messengers [18]
- Paracrine or autocrine
- Exogenous FA can increas levels of the intracellular signals linked to activation of the GF receptors [18]
- FA may promote membrane backbones
- Type of dietary fat influence the composition of FAs of pancreatic membranes [18]
- High-fat diet
- Induce inflammation in K-Ras (G12D) mouse models
- Was shown to enhance tumor promotion [18]
- Enhanced pancreatic tumorigenesis by:
- Via + expression of genes encoding regulators of FA uptake and oxidation
- + metabolic rates in FA
- + energy uptake if FA
- High-fat diet and obesity
- Strongly associated with the incidence of human pancreatic cancer
Omega-3 MK
- Consumption of omega-3 fatty acids
- Suppressive effect on the progression of breast, prostate and colon cancers [18]
- Omega-3-based fat diet in (EL)-K-Ras transgenic mice that develop pancreatic neoplasia
- Blockage of cell-cycle progression
- Induction of programmed cell death [18]
Omega-6 fatty acids consumption
- Highly correlated with progression of breast, prostate, colon and pancreatic tumors [18]
Gamma delta T cells
- Prevents other tumor-fighting T cells from entering pancreatic tumors
- Infiltrate pancreatic tumors
- Prolific in human PDA tumors
- Cca 40 percent of T cells on average
- Enable pancreatic cancer tumors to grow unchecked
- Unless the gamma delta T cells are blocked, CD4 and CD8 cells are unable to function or thwart cancer growth
Inhibice gamma delta T cells
- CD4 and CD8 cells multiply and actively attack tumors
- Mice harboring pancreatic cancer with fewer than normal gamma delta cells survived nearly a year longer on average than mice with a normal number
Glutamine
- Most abundant amino acid in the cytoplasm
- Primary sources of carbon for:
- ATP production
- Biosynthesis in tumorigenic cells
- In proliferating cells TCA provides:
- ATP
- Intermediate metabolites
- Exit the cycle and become converted primarily to:
- Fatty acids
- Nonessential amino acids (NEAAs)
- Glutamine - one of the major anaplerotic precursors
- Important provider of nitrogen for:
- Generation of nucleotide
- NEAAs
- Hexosamine
- nicotinamide [18]
- Reloading the TCA cycle
- Glutamine needs to be converted to alfa-ketoglutarate
- A central metabolite of glutamine metabolism [18]
- Enzymatic conversion of glutamine can be mediated through:
- 1) canonical anabolic glutamine metabolism:
- By an oxidative deamination reaction
- Catalyzed by the mitochondrial matrix enzyme glutamate dehydrogenase (GLUD1)
- GLUD1 drives glutamine into the TCA cycle
- Promotes tumorigenic cell anabolism [18]
- 2) noncanonical metabolic pathway:
- Conducted by transaminases
- Catalyze transamination
- Transfer of an amino group from glutamate to the corresponding alfa-ketoglutarate [18]
- ‘glutamine addiction' of many cancer cells
- Survival dependent on their glutamine content [18]
- Synthesis of nitrogen-based nucleotides and NEAAs
- Fundamental metabolic step in cancer cell growth
- Donation of amide group from glutamine
- Conversion of glutamine to glutamic acid [18]
- Essential nitrogen donor in:
- 3 autonomous enzymatic reactions in purine synthesis
- 2 reactions of pyrimidine synthesis [18]
- In PDAC cells glutamine seems to:
- Support tumorigenic growth by utilizing the noncanonical pathway [18]
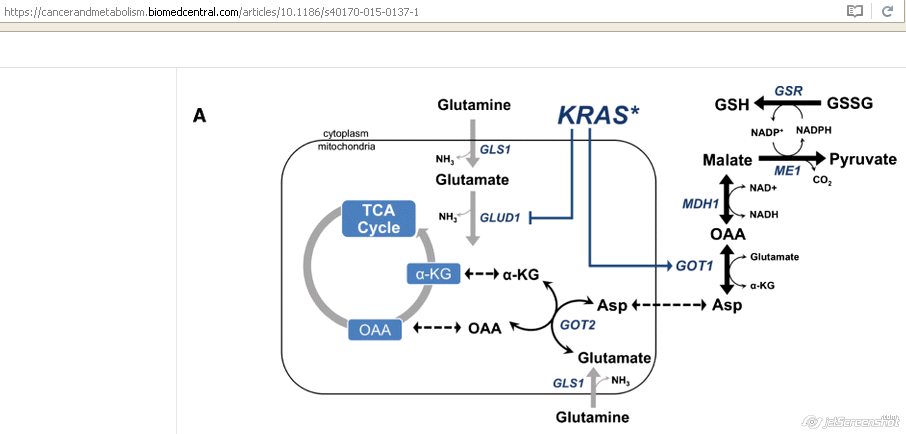
- Glutamine-derived aspartate into the cytoplasm
- Further converted by GOT1 into oxaloacetate
- Into malate
- Finally into pyruvate and NADPH is generated
- Reduction in the NADP+/NADPH ratio
- Enables PDAC cells to maintain their redox homeostasis
- Support cell proliferation [18]
- Cancers use glutamine to generate antioxidants
- This pathway is unique to pancreatic cancer [22]
Silencing of glutaminase
- Generates glutamate from glutamine
- Significantly reduce PDAC cell growth
- Addition of glutamate to the media restored cell growth
GLUD1 inhibitors
- Epigallocatechin gallate
- GLUD1 shRNA [18]
- Did not affected the cell growth [18]
Alfa-ketoglutarate
- Product of canonical oxidative deamination of glutamate
- Failed to restore pancreatic cancer cell growth
- Supplementation of the end-products of the noncanonical transaminase-mediated glutamine pathway
Alfa-ketoglutarate + NEAA mixture
- Significantly restored proliferation in multiple PDAC lines
Transaminase pan-inhibition
- Growth inhibition by:
- Aminooxyacetate
- Aspartate transaminase – glutamic-oxaloacetic transaminase 1 (GOT1) – was depleted
Growth factors
HK
- First glycolytic enzyme
- Phosphorylates glucose to produce glucose-6-phosphate
Inhibitors of HK
- Interfere with the preparatory phase [18]
hMLH1
H-Ras
- H-RAS transformed mesenchymal stem cells
- Do not depend on increased glycolysis for ATP production during transformation [15]
Cholesterol
- Controlling cholesterol metabolism in pancreatic cancer cells reduces metastasis [19]
- Accumulations of the compound cholesteryl ester in human pancreatic cancer specimens and cell lines
- Link between cholesterol esterification and metastasis
- Esterification allows cholesterol to be stored in cells
- Excess quantities of cholesterol result in cholesteryl ester being stored in lipid droplets within cancer cells [19]
- The accumulation of cholesteryl ester
- Controlled by an enzyme called ACAT-1
- Higher expression of the enzyme = poor survival rate for patients
Depletion of cholesterol esterification
- Significantly reduced pancreatic tumor growth and metastasis in mice [19]
Avasimibe - potent inhibitor of ACAT-1
- Previously developed for treatment of atherosclerosis
- Pancreatic cancer cells were much more sensitive to ACAT-1 inhibition than normal cells
- Reduced the accumulation of cholesteryl ester [19]
- Decrease of the number of lipid droplets
- Reduction of cholesteryl ester in the lipid droplets
- Avasimibe acted by blocking cholesterol esterification
- Did not induce weight loss
- no apparent organ toxicity in the liver, kidney, lung and spleen [19]
- Blocking storage of cholesteryl ester
- Causes cancer cells to die
- Damage to the endoplasmic reticulum [19]
- Avasimibe mouse treatment for four weeks
- Remarkably suppressed tumor size
- Largely reduced tumor growth rate
- A much higher number of metastatic lesions in lymph nodes were detected in the control group than the avasimibe-treated
- Each mouse in the control group showed at least one metastatic lesion in the liver.
- Only three mice in the avasimibe treated group [19]
INK4A/ARF
Loss of tumor suppressors INK4A/ARF
Ki67
Suppression of Ki67
- Consistent with the established role for CDK4/6
- Increase in mitochondria [17]
- Observed in cell lines
KRAS
- KRAS - challenging drug target
- Point mutations are known to be involved in pancreatic oncogenesis [13]
- Ki-ras codon 12 mutations were found in 14 of 20 (70%) pancreatic cancers
- Significantly higher incidence of c-Ki-ras than p53 gene mutations [14]
- Vital role in controlling tumor metabolism
- Critical role in pancreatic ductal adenocarcinoma initiation [15]
- >90% of cases [15]
- Mutation in K-Ras genes - detectable also in the plasma DNA of patients with pancreatic cancer
- Often correlated with more advanced stage disease
- Often detected in pancreatic intraepithelial neoplasia (PanIN) and PDAC
- Mutations in K-Ras alone seem to be insufficient for transformation of the pancreatic tissue to PDAC [18]
- Endogenous expression of active K-Ras(G12D) in progenitor cells that reside in the mouse pancreas
- Frequent development of highly proliferative PanIN
- Infrequent progression to more invasive metastatic adenocarcinomas [18]
- Transfection of K-Ras(G12V) into human pancreatic duct epithelial cells derived from normal human pancreas
- Resulted in only moderately aberrant phenotypes [18]
Activation of the Ki-ras oncogene - Oncogenic KRAS
- Can induce senescent-like growth arrest state in cells [17]
- Mediated by p16ink4a encoded by CDKN2A that blocks the activity of:
- CDK4/Cyclin D complexes
- CDK6/Cyclin D complexes [17]
- This leads to:
- Suppression of RB phosphorylation
- Concomitant inhibition of cell-cycle progression
- Through the suppression of E2F-mediated transcription [17]
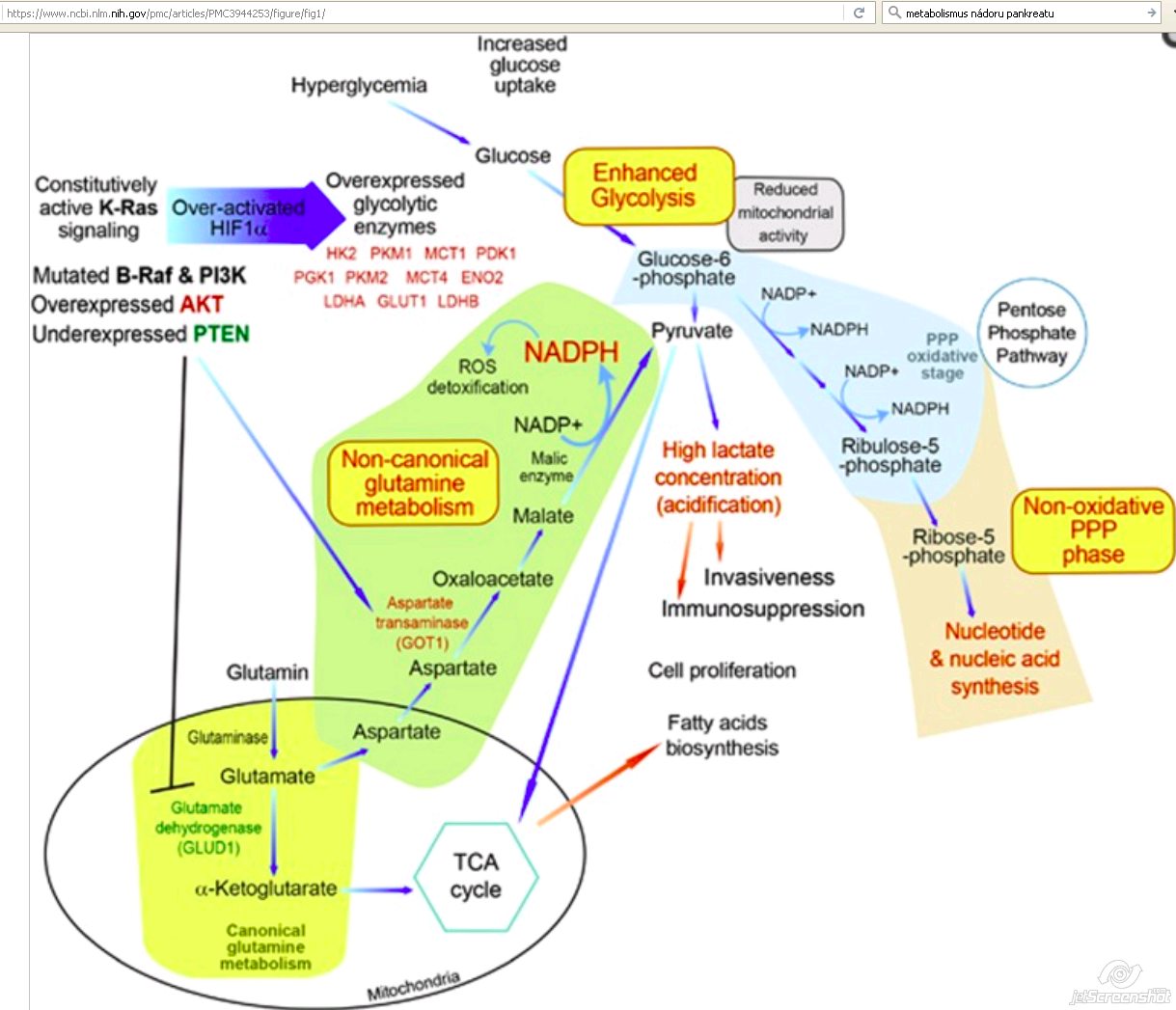
- Stimulate of glucose uptake
- + hexosamine biosynthesis
- + pentose phosphate pathways (PPP) [15]
- + nonoxidative PPP
- Decoupling ribose biogenesis from NADP/NADPH-mediated redox control [15]
- Promotes ribose biogenesis [15]
- Promotes macropinocytosis [17]
- Constitutive KrasG12D signaling:
- Drives uncontrolled proliferation
- Enhances survival of cancer cells
- Via activation of downstream signaling pathways:
- MAPK
- PI3K-mTOR [15]
- Increased anabolic needs of enhanced proliferation [15]
- + G6P
- + F6P
- + pentose phosphate pathway (PPP) [15]
- + R5P for DNA/RNA biosynthesis [15]
Inhibition of KRAS
- Many agents that target KRAS signaling suppress metabolism [17]
- KrasG12D extinction was accompanied by:
- A significant drop in
- Glucose-6-phosphate (G6P)
- Fructose-6-phosphate (F6P)
- Fructose-1,6-bisphosphate (FBP) [15]
- With minimal changes to the remaining components in glycolysis
- Decreased glucose uptake
- Decreased lactate production
- Downregulation of the
- Glucose transporter (Glut1/Slc2a1)
- Hk1, Hk2, and Pfkl, Ldha [15]
- Rapid reduction in SMA-positive pancreatic stellate cells
- Correlation between stromal SMA positivity and poor prognosis in PDAC patients [15]
- KrasG12D inactivation was not accompanied by:
- Significant alterations to TCA cycle intermediates
- Glutamine is the major carbon source for the TCA cycle in the KrasG12D-driven PDAC cells [15]
- Reduced glutathione (GSH) and oxidized glutathione (GSSG) levels
- Are regulated by NADPH
- Were not significantly altered by KrasG12D inactivation [15]
- Suppressed mTOR signaling [15]
- Rapamycin treatment
- Did not induce extensive changes in glucose metabolism [15]
Subpopulation of dormant tumour cells surviving oncogene ablation
- Responsible for tumour relapse
- Features of cancer stem cells
- Relies on oxidative phosphorylation for survival [23]
- Prominent expression of genes governing:
- mitochondrial function
- Autophagy
- Lysosome activity
- mitochondrial respiration [23]
- Decreased dependence on glycolysis for cellular energetics [23]
- High sensitivity to oxidative phosphorylation inhibitors
- Can inhibit tumour recurrence
KRAS inhibition + inhibition of mitochondrial respiration
LDHA
- Critical function as a generator of glycolysis in cancer cells
- Catalyzes the reversible conversion reaction of pyruvate to lactate
- Utilizes NADH as a co-factor for catalyzing its reaction
- NAD+ is a vital electron acceptor
- Sustains the glycolytic pathway through its reaction with GAPDH;51 [18]
Inhibitors of LDHA
- Inhibition of LDHA has a critical effect on glycolysis [18]
- Ratio of NADH to NAD+ is increased as result of LDHA inhibition [18]
- Inhibition of its activity in a mouse pancreatic cancer model
- Glycolysis shutdown
- ATP reduction
- Significant induction of oxidative stress
- Glycolytic blockade culminates in tumor growth inhibition of pancreatic cells [18]
Malate
- An intermediate in the TCA cycle
- Can be converted into pyruvate by malic enzyme
- With the production of NADPH
- Reducing equivalent
- Used to generate reduced glutathione
- Allowing cancer cells greater tolerance to free radical-induced damage [21]
MAPK
Aktivace MAPK/ERK signaling
- Enhance HIF-1? transcriptional activity [18]
MAPK inhibition
- Recapitulated the KrasG12D inactivation and induced metabolite changes in the:
- Glycolysis
- HBP
- Nonoxidative PPP pathways [15]
MEK
- Predominantly required for the maintenance of glycolytic metabolism [17]
MEK inhibition
- Significantly decreased expression of:
- Glycolytic genes Glut1, Hk1, Eno1, Ldha), the rate-limiting HBP gene (Gfpt1)
- Nonoxidative PPP gene (Rpia) [15]
- Augmented the impact of CDK4/6 inhibition on oxidative metabolism
- + mitochondrial mass
- + cellular complexity
- + OCR
- Selectively inhibited glycolysis [17]
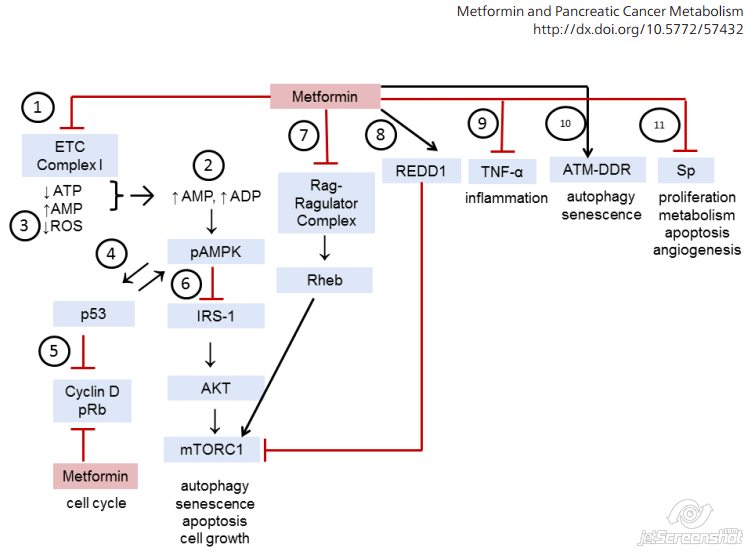
mTOR
- Mechanistic target of rapamycin (mTOR)
- (formerly mammalian target of rapamycin
- Kinase that in humans
- Encoded by the MTOR gene
- Member of the phosphatidylinositol 3-kinase-related kinase family of protein kinases
- Core component of two distinct protein complexes:
- MTOR complex 1
- MTOR complex 2 [27]
- Functions as a serine/threonine protein kinase that regulates:
- Cell growth
- Cell proliferation
- Cell motility
- Cell survival
- protein synthesis
- Autophagy
- Transcription [27]
- As a tyrosine protein kinase that promotes:
- The activation of insulin receptors
- Insulin-like growth factor 1 receptors [27]
- MTORC2
- Control and maintenance of the actin cytoskeleton [27]
- Regulated by:
- PI3K/AKT/TSC pathway
- Amino acid availability
- Depletion of amino acids
- Blocked the induction of MTOR activity with CDK4/6 inhibition [17]
- Lysosomes
- Appropriate milieu of regulatory proteins [17]
- MTOR Complex 1 (mTORC1) is composed of:
- MTOR
- Regulatory-associated protein of MTOR (Raptor)
- Mammalian lethal with SEC13 protein 8 (MLST8)
- Non-core components PRAS40 and DEPTOR [27]
- Activity of mTORC1 is regulated by:
- Rapamycin
- Insulin
- Growth factors
- Phosphatidic acid
- Certain amino acids and their derivatives
- L-leucine
- ß-hydroxy ß-methylbutyric acid [27]
- Mechanical stimuli
- Oxidative stress [27]
- MTOR Complex 2 (mTORC2) is composed of:
- MTOR
- Rapamycin-insensitive companion of MTOR (RICTOR)
- MLST8
- Mammalian stress-activated protein kinase interacting protein 1 (mSIN1) [27]
- MTORC2 has been shown to function as:
- Important regulator of the actin cytoskeleton via stimulation:
- F-actin stress fibers
- Paxillin
- RhoA
- Rac1
- Cdc42
- protein kinase C alfa (PKC alfa) [27]
- MTORC2 also
- Phosphorylates the serine/threonine protein kinase Akt/PKB on serine residue Ser473
- Affecting metabolism and survival
- Stimulates Akt phosphorylation on threonine residue Thr308 by PDK1
- Full Akt activation [27]
- Tyrosine protein kinase activity
- Phosphorylates the insulin-like growth factor 1 receptor (IGF-IR)
- Phosphorylates insulin receptor (InsR) on the tyrosine residues Tyr1131/1136 and Tyr1146/1151
- Full activation of IGF-IR and InsR [27]
Aktivace mTOR
- Stimulate metabolism leading to cell-cycle progression
- Antagonistic to the cytostatic effect of CDK4/6 inhibition
- Stimulován CDK 4/6 inhibitory ! [17]
- Induction of multiple gene expression programs:
- Glycolysis
- Lysosome biogenesis
- Fatty acid metabolism
- PPAR signaling [17]
- As a consequence of amino acid availability and lysosomal localization
- CDK4/6 inhibition yielded:
- Rapid accumulation of lysosomes
- Increased amino acid pools
- Energetic feedforward loop
- MTOR activity is required for metabolic reprogramming induced by CDK4/6 inhibition [17]
- MTORC1 initiates a phosphorylation cascade activating the ribosome
- Proportion of damaged proteins is enhanced [27]
- Over-activation of mTOR signaling
- Initiation and development of tumors
- Deregulated in many types of cancer
- Constitutive activation or mTOR:
- Mutations in tumor suppressor PTEN gene
- PTEN phosphatase negatively affects mTOR signalling
- Effect of PI3K, an upstream effector of mTOR [27]
- Increased activity of PI3K
- Increased akcitivity of Akt [27]
- Overexpression of downstream mTOR effectors:
- 4E-BP1
- S6K
- EIF4E [27]
- Mutations in TSC protein
- Inhibits the activity of mTOR
- Tuberous sclerosis complex [27]
- Increasing mTOR activity
- Drive cell cycle progression
- Increase cell proliferation
- protein synthesis [27]
- Tumor growth
- By inhibiting autophagy [27]
- Constitutively activated mTOR
- Supplying carcinoma cells with oxygen and nutrients
- Increasing the translation of HIF1A
- Supporting angiogenesis
- Activation of glycolytic metabolism
- Akt2, a substrate of mTORC2, upregulates expression of the glycolytic enzyme PKM2
- Contributing to the Warburg effect [27]
- MTOR
- Implicated in the failure of a 'pruning' mechanism of the excitatory synapses
- In autism spectrum disorders [27]
mTOR inhibice
- Fully suppressed metabolism
- Yielded apoptosis
- Yielded suppression of tumor growth in xenograft models [17]
- Cooperated with CDK4/6 inhibition
- Suppression of ROS scavenging
- BCL2 antagonists [17]
- MTOR and MEK inhibitors
- Potently cooperate with CDK4/6 inhibition in eliciting cell-cycle exit [27]
- Rapamycin (sirolimus)
- Arrests fungal activity at the G1 phase of the cell cycle
- In mammals, it suppresses the immune system
- By blocking the G1 to S phase transition in T-lymphocytes
- Used as an immunosuppressant following organ transplantation [27]
- Rapamycin inhibits mTORC1
- Most of the beneficial effects of the drug
- Life-span extension in animal studies
- More complex effect on mTORC2
- Inhibiting it only in certain cell types under prolonged exposure [27]
- Disruption of mTORC2 produces:
- Diabetic-like symptoms of decreased glucose tolerance
- Insensitivity to insulin [27]
- Inhibition of mTORC1 and mTORC2 by PP242 [2-(4-Amino-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-1H-indol-5-ol]
- Leads to autophagy or apoptosis [27]
- Inhibition of mTORC2 alone by PP242
- Prevents phosphorylation of Ser-473 site on AKT
- Arrests the cells in G1 phase of the cell cycle [27]
- Genetic reduction of mTOR expression
- In mice significantly increases lifespan [27]
- Decreased TOR activity
- Found to increase life span [27]
- Some dietary regimes cause lifespan extension by decreasing mTOR activity
- Caloric restriction
- methionine restriction
- leucine (which are potent activators of mTOR)
- Administration of leucine into the rat brain
- Decrease food intake and body weight via activation of the mTOR pathway in the hypothalamus [27]
- ATP sensitive AMPK, the mTOR pathway is inhibited
- ATP consuming protein synthesis is downregulated
- Disruption of mTORC1 directly
- Inhibits mitochondrial respiration [27]
- Decreased mTOR activity
- Upregulates glycolysis
- Up regulates removal of dysfunctional cellular components via autophagy [27]
- Epigallocatechin gallate (EGCG), caffeine, curcumin, and resveratrol [27]
- Temsirolimus
- Everolimus
- Ridaforolimus
Combined CDK4/6 and MTOR inhibition
- Suppression of both glycolytic and oxidative functions [17]
- Suppressed senescence
- Resulted in the induction of apoptotic cell death [17]
- Could not be reversed by supplementation with methyl-pyruvate or alpha-ketoglutarate [17]
- MTOR inhibitors
- Restricted glycolytic metabolism and oxidative phosphorylation induced by CDK4/6 inhibition
- + cell death
- + suppression of tumor growth [17]
MEK + MTOR inhibition + CDK4/6 inhibition
Nrf2
- ‘detox’ protein from ROS
- In pancreatic cancer cells a faulty version of a gene called K-Ras sparked an unexpected upsurge in production of the antioxidant Nrf2
- ‘companion protein’ recruited by K-Ras
Inhibice Nrf2 signalling pathway
- Decreased the development of both pancreatic and lung tumours [28]
p16 ink4a
Loss of p16ink4a
- In pancreatic ductal adenocarcinoma (PDA )
Náhrada funkce
- Highly selective drugs that phenocopy features of p16ink4a function
- Would be expected to have potency in PDA
- Such drugs have some degree of effect in established PDA cell lines
- Resistance can develop quickly
- Necessitating the use of combination therapeutic approaches [17]
p53 tumor suppressor gene - TP53
- Critical role in cell cycle regulation
- Nuclear transcription factor
- Point mutations in the p53 gene have been observed [13]
- P53 mutations were found in 5 of 20 pancreatic cancers
- Three of 14 primary tumors, two of six metastatic tumors [13]
- 29 of 71 (41%) tumors showed mutations of the p53 gene [14]
- Majority were missense point mutations
- Primarily within the evolutionary conserved domains (62%) [14]
- 1/3 of the carcinomas both Ki-ras codon 12 and p53 gene mutations [14]
Inactivation of the p53 tumor suppressor gene - TP53
- P53 mutations correlated with
- Distant metastasis (p < 0.05)
- Survival (p < 0.05) [14]
- Seem to be associated with a metastatic phenotype
- Possibly acquired during tumor progression [14]
PALB2
PHGDH
- A rate-limiting enzyme
- Divert 3-phospho-glycerate (a glycolytic intermediate) into the serine biosynthesis pathway
Amplification/overexpression of PHGDH
- Facilitates tumor growth in certain contexts [15]
PI3K-AKT
- PIK3Ca - the gene encoding PI3K
Stimulace PI3K
- Mutations during tumorigenesis of pancreatic cells
- Cca 10% of pancreatic cancer precursors harbor activating mutations in this gene [18]
Amplification / overactivation of Akt2 kinase
- Direct signaling target of PI3K
- 60% of pancreatic cancers [18]
Aktivace PI3K/Akt signaling pathway
- Strongly associated with:
- Elevated expression levels and translocation to the cellular membrane of the GLUT1 [18]
- Stimulation of phosphofructokinase enzymatic activity
- mitochondrial localization of the glycolytic enzymes HK1 and HK2 [18]
- Stabilization of HIF-1? protein [18]
Inhibition of PI3K-AKT signaling (BKM120)
- Did not exhibit a significant impact on iKras-directed tumor metabolism [15]
PPP (or phosphogluconate pathway)
- Biphasic cytosolic process
- Utilizes glucose-6-phosphate
- Biochemical alternative to glycolysis
- Anabolic process
1st phase = ‘oxidative' PPP
- Enzymes involved were not altered upon KrasG12D extinction [15]:
- G6pd
- Pgls
- Pgd
- Enzymatic activity of G6pd
- The rate-limiting step for the oxidative arm
- Produces NADPH - a reducing agent in biosynthetic reactions:
- Biogenesis reactions
- Rebuild macromolecules
- As an antioxidative metabolite in the detoxification of ROS
- Via regeneration of reduced glutathione
- Providing protection against hostile microenvironments
- Glutathione reductase together with NADPH-generating pathways and glutathione
- Provides cellular defense system against oxidants
- Highly expressed in pancreatic islet cells [18]
- K-Ras-driven accelerated glycolytic flux in pancreatic tumor cells
- Reprogrammed to bypass the oxidative NADPH biosynthesis phase
- Instead robustly enhances the nonoxidative branch [18]
2nd phase = ‘nonoxidative' PPP
- Importance of the reversible nonoxidative PPP phase in pancreatic tumor cells [18]
- Produces 5-carbon sugars
- Primary intermediates in the synthesis of nucleotides and nucleic acids [18]
- Enzymes that regulate carbon exchange reactions
- Significantly decreased by KRAS inhibition [15]
Stimulation of nonoxidative PPP
- By mutated K-Ras
- + generating ribose-5-phosphate [18]
- + synthesis of nucleic acids [18]
- Linking K-Ras directly to DNA biosynthesis [18]
Inhibition of the Nonoxidative PPP
- Suppresses KrasG12D-Dependent Tumorigenesis [15]
Knockdown of either Rpia or Rpe
- Significantly reduced the flux of 14C1-labeled glucose into DNA/RNA [15]
- In high-glucose (11 mM)
- Moderately suppresses the clonogenic activity of iKras p53L/+ tumor cells
- In low-glucose containing media (1 mM) [15]
- Inhibitory effect is dramatically enhanced [15]
PRSS1
PTEN - Phosphatase and tensin homolog encodes phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase
- Antagonist signaling factor vůči RAS
- In pancreatic cancers is lost or significantly reduced [18]
- Tumor suppressor
- Mutated in a large number of cancers at high frequency
- Preferentially dephosphorylates phosphoinositide substrates
- Negatively regulates intracellular levels of phosphatidylinositol-3,4,5-trisphosphate in cells [29]
- Negatively regulating Akt/PKB signaling pathway
- Phosphatase to dephosphorylate phosphatidylinositol (3,4,5)-trisphosphate (PtdIns (3,4,5)P3 or PIP3) [29]
- Specifically catalyses the dephosporylation of the 3` phosphate of the inositol ring in PIP3
- Biphosphate product PIP2 (PtdIns(4,5)P2)
- Results in inhibition of the AKT signaling pathway [29]
- Weak protein phosphatase activity
- Role as a tumor suppressor
- Regulation of the cell cycle [29]
- Numerous reported protein substrates for PTEN
- IRS1
- Dishevelled [29]
- Targets for drugs:
- OncomiR
- MIRN21 [29]
Inaktivace PTEN
- Increased cell proliferation
- Reduced cell death [29]
- PTEN mutation also causes a variety of inherited predispositions to cancer [29]
- Defective protein is unable to stop cell division or signal abnormal cells to die
- Mutations in the PTEN gene
- PTEN hamartoma tumor syndromes [29]
- PTEN deletion mutants have recently been shown to allow nerve regeneration in mice
- PTEN inhibitors
- Bisperoxovanadium compounds
- Neuroprotective effect after CNS injury [29]
PTEN agonists
- Rapamycin
- Sirolimus
- Temsirolimus [29]
Ras oncogene
- Genes
- H-Ras, N-Ras, K-Ras
- Encoding 4 highly homologous small GTPase Ras proteins:
- H-Ras, N-Ras, K-Ras4A, K-Ras4B
- Larger Ras-related GTPase protein superfamily
- Molecular switches
- Inactive (GDP-bound) conformations
- Active (GTP-bound) conformations
Aktivace RAS
- Extracellular signals
- Plasma membrane-bound receptor tyrosine kinases
- Activation of Ras guanine nucleotide exchange factors (GEFs)
- GTP-bound state = affinity of Ras for a multitude of intracellular factors is increased
- Diversity of signal transduction cascades:
- PI3K-Akt
- Raf-Mek1/2-Erk1/2
- RalGEF
- Rho/Rac
- Activated Ras signals
- Coming from the plasma membrane
- Carried over the intracellular compartments by a variety of molecular mediators
- Target in the nucleus
- Activity of transcription factors like:
- Myc
- NF-?B
- E2F
- HIF-1?
- AP-1
- C-Jun [18]
- Cell survival
- Migration
- Cell cycle
- Metabolism
- Differentiation [18]
- Promote glycolysis [15]
- Ras pathway stimulate
- Cellular glucose uptake
- Metabolic rate
- Overcoming the capacity of the cell to utilize mainly glucose
- Secrete excess metabolites of the glycolytic pathway
- Lactic acid (Warburg effect) [18]
Mutace aktivující RAS onkogen
- >90% of patients with pancreatic cancer K-Ras mutation on chromosome 12p (codons 12, 13 and 61) [18]
- Constitutively active Ras pathways:
- Maintain the carcinogenic phenotype
- Alters the metabolic pathways
- Promote rapid progression of pancreatic tumors [18]
Ras inhibitors
- Trans-farnesylthiosalicylic acid (also known as Salirasib) [18]
- Specifically target the active form of Ras (Ras-GTP)
- Potent antiproliferative effects
- Melanoma
- Merkel cell carcinoma
- LNCaP
- CWR-R1
- Panc-1
- MIA PaCa-2 pancreatic cancers [18]
- Profound anti-oncogenic effects in glioblastoma multiforme cells
- By degradation of HIF-1?
- Blocked glycolysis
- Triggered severe energetic crises [18]
- Ras inhibition, through glycolysis shutdown (2-DG)
- Can impose in pancreatic tumors
- Glycolysis emerges as one of the central metabolic modules to which pancreatic tumor cells become addicted [18]
RB
- Bind to mitochondria
- Regulate apoptotic functions [17]
- Increase in mitochondrial mass was dependent on RB [17]
RB loss
- Increased glutamine utilization [17]
- In fibroblastic models [17]
- Knockdown of RB1
- Partially reverted the accumulation of mitochondria [17]
- Selectively associated with a diminution of oxidative phosphorylation [17]
- Increased sensitivity to mitochondrial poisons [17]
RB activation
- B activation by CDK4/6 inhibition
- Activates mitochondrial function [17]
- Cells are less sensitive to glucose withdrawal [17]
- Less sensitive to mitochondrial poisons [17]
Ribose and glutamin
- PDA cells generate the bulk of the ribose
- Used for de novo nucleotide biosynthesis
- Via non-oxidative arm of the pentose phosphate pathway [16]
- Bypasses the nicotinamide adenine dinucleotide phosphate (NADPH)
- Generating oxidative arm [16]
- To compensate for this rewiring utilize glutamine through:
- GLS1 (mitochondrial glutaminase)-
- GOT2 (mitochondrial glutamate oxaloacetate transaminase 2)
- GOT1 (cytoplasmic glutamate oxaloacetate transaminase 1)-dependent pathway
- To support cellular redox balance in the face of rapid proliferation and growth [16]
- Genetic inhibition of enzymes in this pathway is profoundly growth inhibitory in PDA
- Does not result in the induction of a cytotoxic response [16]
SCD1
- Endoplasmic reticulum-bound protein
- Encoded by the SCD1 and SCD5 genes in humans
- Highly expressed in:
- Liver and adipose tissue (SCD1)
- Brain and the pancreas (SCD5) [21]
- Saturation index 18:0 to 18:1 n-9 ratio
- Marker for SCD activity
- Associated to cancer risk [21]
SCD inhibition
- Chemical or genetic
- Inhibition of cancer cell proliferation and/or death [21]
- Modulating lipid metabolism and signaling processes
- Cancer cell replication and anchorage-independent growth [21]
SMAD4
Loss of tumor suppressors SMAD4
STK11
Substrates
- Starving cells of either glutamine or glucose significantly reduced viability [17]
- Pretreatment with CDK4/6 inhibitors
- Protected selectively against the effect of acute glucose withdrawal [17]
Vyživování okolními buňkami
- Pancreatic cancer cells grow by instructing neighboring cells to provide them with nutrients
- Cells from the tumor microenvironment degrade their own proteins
- Supply the cancer cells with the resulting amino acids [20]
- Interactions with stromal cells
- Thick stroma protects pancreatic cancer cells from exposure to chemotherapy drugs
Alanine
- Using alanine as a main energy source
- Allows “the cancer cells to utilize glucose for building DNA and RNA [20]
Pancreatic stellate cells (PSCs)
- Star-shaped cells
- Secrete structural proteins
- Abundant in the stroma
- PDAC cells encourage PSC growth
- PSCs, in turn, can promote PDAC growth [20]
- Amino acids secreted by PSCs are absorbed by PDAC cells:
- Aspartate
- Alanine
- Only alanine stimulated mitochondrial metabolism in PDAC cells [20]
- PSCs may produce extra alanine through autophagy
- Degrades superfluous, damaged, and toxic molecules into basic units [20]
- Co-cultured PDAC and PSC cells together
- Autophagy increased in the PSCs [20]
- PSCs required essential autophagy genes to secrete alanine and enhance PDAC metabolism
- PSC culture medium enhanced PDAC cell growth in low-nutrient conditions
- Effect was dependent on autophagy in PSCs [20]
- Mice tumors from PDAC cells implanted along with PSCs that lacked autophagy-related genes
- Grew slower
- Were less lethal [20]
Warburg effect
- Most cancer cells, also in pancreas tumors
- Even under nonhypoxic conditions
- Predominantly utilize cytosolic aerobic glycolysis and lactate fermentation
- Rather than mitochondrial oxidative phosphorylation of pyruvate [18]
- + oxidative glycolytic enzymes expression:
- Hexokinase 2 (HK2)
- Phosphoglycerokinase 1
- Pyruvate dehydrogenase kinase isozyme 1 (PDK1)
- Lactate dehydrogenase A and B (LDHA, LDHB)
- Enolase 2 (ENO2)
- Pyruvate kinase muscle (PKM1 and PKM2)
- Glucose and lactate transporters
- PDK1
- LDHA, LDHB
- Glucose transporter 1 (GLUT1)
- Monocarboxylate transporters 1 and 4 (MCT1 and MCT4) [18]
Glycolytic ATP production is advantageous for the cancer cells:
- Inconstant oxygen diffusion
- Deleterious for normal cells
- Lactic and bicarbonic acids
- Major acidity buffer
- Preferentially promotes the invasiveness of these cells [18]
- Correlation between tumor burden and high lactate levels
- Intracellular accumulation of lactic acid links the antitumorigenic immune response
- Activated cytotoxic tumor-infiltrating T lymphocytes rely on glycolysis
- Tightly dependent on efficient secretion of lactic acid
- Increased concentrations of lactate in the tumor environment lead to a decline in the intracellular/extracellular lactate gradient
- Blocking lactate secretion and consequently the entire metabolism in T cells
- Immunosuppression in the pancreatic tumor niche is strongly increased
- Inhibition of T-cell proliferation
- Suppression of their cytokine production
- Increase in ENO1-specific regulatory T cells (Tregs)
- Detected in pancreatic cancer tumor cells
- High levels of the key glycolytic enzyme ENO1
- Efficiently impose immunosuppression in vitro
- Suppressing the proliferative response of ENO1-specific effector T cells [18]
- Possible role for Tregs in stimulating pancreatic cancer progression [18]
- Tumor cells utilize glycolytic intermediates to fuel anabolic processes:
- Pyruvate
- Alanine aminotransferase synthesizes alanine and malate [18]
- Can enter the tricarboxylic acid (TCA) cycle
- Leading to the export of acetyl coenzyme A (acetylCoA) from the mitochondrial matrix to the cytosol and thus making acetylCoA available for synthesis:
- Of fatty acids
- cholesterol
- Isoprenoids [18]
- Glucose 6-phosphate
- Converted by phosphoglucomutase [18]
- glycogen
- By phosphorylase [18]
- Dihydroxyacetone phosphate
- To triacylglyceride + phospholipid [18]
- Expression of fatty acid synthase
- Catalyze the synthesis of long-chain fatty acids
- From nicotinamide adenine dinucleotide phosphate (NADPH), acetylCoA and malonyl coenzyme A
- Correlate with an advanced stage of pancreatic cancer [18]
Inhibice oxidativní glykolýzy
- Potent anti-glycolytic effect of everolimus, a rapamycin analog, on pancreatic Panc-1 human cancer cells
- Gradual increase in expression of miR-143
- Consequently targets and reduces expression of the preparatory enzyme HK2 [18]